Why care about open (data) science?
Overview
Teaching: 40 min
Exercises: 10 minQuestions
What is Open Science?
What is Open and Reproducible Research Practices?
Objectives
To understand the importance to share data and code
To value code and data for what they are: the true foundations of any scientific statement.
To promote good practices for open & reproducible science
The Crisis of Confidence
The crisis of confidence poses a general problem across most empirical research disciplines and is characterized by an alarmingly low rate of key findings that are reproducible (e.g., Fidler et al. 2017; Open Science Collaboration, 2015; Poldrack et al., 2017; Wager et at., 2009). A low reproducibility rate can arise when scientists do not respect the empirical cycle. Scientific research methods for experimental research are based on the hypothetico-deductive approach (see e.g., de Groot, 1969; Peirce, 1878), which is illustrated in Figure 1.
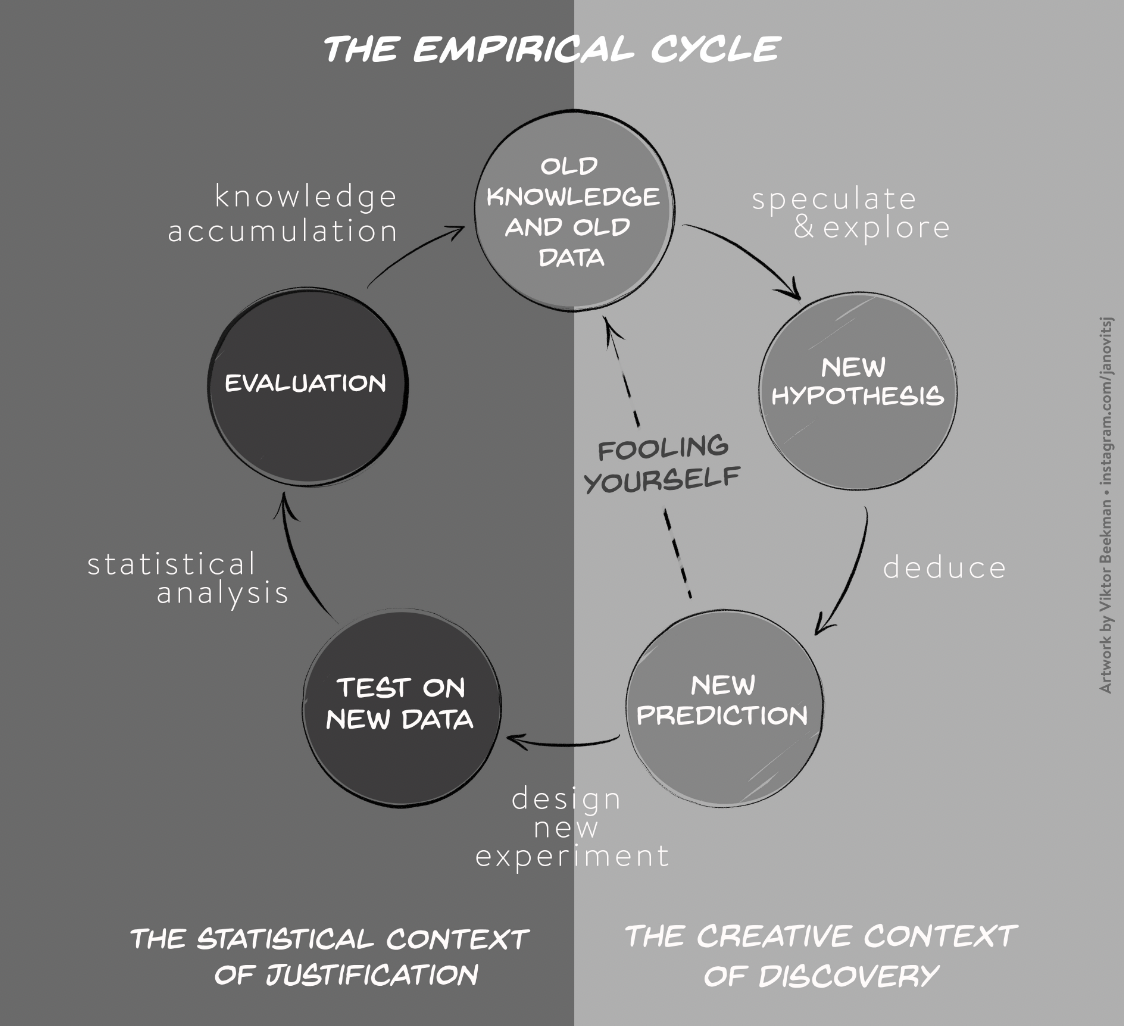
Figure 1. The two stages of the empirical cycle; after the initial stage of creative discovery and hypothesis generation (illustrated in the right panel), researchers test their hypotheses in a statistical context of justification (illustrated in the left panel). However, scientists fool themselves, if they test their new predictions on old knowledge and old data (dotted line).”
The empirical cycle suggests that scientists initially find themselves in “the creative context of discovery”, where the primary goal is to generate hypotheses and predictions based on exploration and data-dependent analyses. Subsequently, this initial stage of discovery is followed by “the statistical context of justification”. This is the stage of hypothesis-testing in which the statistical analysis must be independent of the outcome. Scientists may fool themselves whenever the results from the creative context of discovery with its data-dependent analyses are treated as if they came from the statistical context of justification. Since the selection of hypotheses now capitalizes on chance fluctuations, the corresponding findings are unlikely to replicate.
This suggests that the crisis of confidence is partly due to a blurred distinction between statistical analyses that are pre-planned and post-hoc, caused by the scientists degree of freedom in conducting the experiment, analyzing the data, and reporting the outcome. In a research environment with a high degree of freedom it is tempting to present the data exploration efforts as confirmatory (Carp, 2013). Kerr (1998, p. 204) attributed this biased reporting of favorable outcomes to an implicit effect of a hindsight bias: “After we have the results in hand and with the benefit of hindsight, it may be easy to misrecall that we had really ‘known it all along’, that what turned out to be the best post hoc explanation had also been our preferred a priori explanation.”
To overcome the crisis of confidence the research community must change the way scientists conduct their research. The alternatives to current research practices generally aim to increase transparency, openness, and reproducibility. Applied to the field of ecology, Ellison (2010, p. 2536) suggests that “repeatability and reproducibility of ecological synthesis requires full disclosure not only of hypotheses and predictions, but also of the raw data, methods used to produce derived data sets, choices made as to which data or data sets were included in, and which were excluded from, the derived data sets, and tools and techniques used to analyze the derived data sets.” To facilitate their uptake, however, it is essential that these open and reproducible research practices are concrete and practical.
Open and Reproducible Research Practices
In this section, we focus on open and reproducible research practices that researchers can implement directly into their workflow, such as data sharing, creating reproducible analyses, and the preregistration of studies.
Data Sharing
International collaboration is a cornerstone for the field of ecology and thus the documentation, and archiving of large volume of (multinational) data and metadata is becoming increasingly important. Even though many scientists are reluctant to make their data publicly available, data sharing can increase the impact of their research. For instance, in cancer research, studies for which data were publicly available received higher citation rates compared to studies for which data were not available (Piwowar, Day, & Fridsma, 2007). This is due to the fact that other researchers can build directly on existing data, analyze them using utilize novel statistical techniques and modelling tools, and mine them from new perspectives (Carpenter et al., 2009).
Reproducibility of Statistical Results
One of the core scientific values is reprodicibility. The reproducibility of experimental designs and methods allows the scientific community to determine the validity of alledged effects.
The benefit of publishing fully reporducible statistical results (including the reporting of all data preprocessing steps) is that collaborators, peer-reviewers, and independent researchers can repeat the analysis –from raw data and to the creation of relevant figures and tables– and verify the correctness of the results. Scientific articles are not free from typographical mistakes and it has been shown that the prevalence for statistical reporting errors is shockingly high. For instance, Nuijten et al. (2015) examined the prevalence of statistical reporting errors in the field of psychology and found that almost 50% or all psychological articles papers contain at least one error. These reporting errors can lead to erroneous substantive conclusions and influence, for instance, the results of meta-analyses. Most importantly, however, is that these errors are preventable. Through tools, such as git and RMarkdown, researchers can automate their statistical reporting and produce fully reproducible research papers.
Preregistration and Registered Reports
A blurred distinction between statistical analyses that are pre-planned and post-hoc causes many researchers to (unintentionally) use questionable research practices to produce significant findings (QRPs; John, Loewenstein, & Prelec, 2012). The most effective method to combat questionable research practices is preregistration, a procedure to curtail scientists’ degrees of freedom (e.g., Wagenmakers & Dutilh, 2016. When preregistering studies, scientists commit to an analysis plan in advance of data collection. By making a sharp distinction between hypothesis generating and analyzing the data, preregistration eliminates the confusion between exploratory and confirmatory research.
Over the last years, preregistration has quickly gained popularity and has established itself over several platforms and formats. Scientists can now choose to preregister their work either independently —for instance on platforms like https://asPredicted.org or the Open Science Framework (OSF)— or preregister their studies directly in a journal in the format of a Registered Report as promoted by Chris Chambers (2013). Currently about 200 journals —including Nature: Human Behaviour— accept Registered Reports either as a regular submission option or as part of a single special issue (see https://cos.io/rr/ for the full list).
Preregistration is encouraged in the transparency and openness promotion (TOP) guidelines (Nosek et al., 2015 and represents the standard for the analysis of clinical trials; for instance, in the New England Journal of Medicine —the world’s highest impact journal— the registration of Clinical Trials is a prerequisite for publication.
Challenges
- Data sharing: ethical concerns (share data that harm others, e.g., lowering property values or private data that are collected, for instance, through satellites); Solution = share anonimized data, policies need to be developed
- Preregistration: Loosing the flexibility to adapt analysis plans to unexpected peculiarities of the data; Solution = data blinding, which is standard practice in astrophysics)
- Reproducibility: Additional costs associated with the time it takes to adequatly annotate and archive the code so that independent researchers can understand and reproduce fugures and results; Solution = reproducible workflow, for instance by working in Git and Rmarkdown
Exercise: Reflect on your own reserach!
Have you ever had a problem reproducing your own research or someone else’s research? Why do think some research is irreproducible?
Solution
Read more about reproducibility crisis here.
Three messages
If there are 3 things to communicate to others after this workshop, I think they would be:
1. Data science is a discipline that can improve your analyses
- There are concepts, theory, and tools for thinking about and working with data.
- Your study system is not unique when it comes to data, and accepting this will speed up your analyses.
This helps your science:
- Think deliberately about data: when you distinguish data questions from research questions, you’ll learn how and who to ask for help
- Save heartache: you don’t have to reinvent the wheel
- Save time: when you expect there’s a better way to do what you are doing, you’ll find the solution faster. Focus on the science.
2. Open data science tools exist
- Data science tools that enable open science are game-changing for analysis, collaboration and communication.
- Open science is “the concept of transparency at all stages of the research process, coupled with free and open access to data, code, and papers” (Hampton et al. 2015)
- For empirical researchers: transparency checklist (https://eltedecisionlab.shinyapps.io/TransparencyChecklist/)
- Repositories such as the Open Science Framework (https://osf.io/preregistration) offer preregistration templates and the tools to archive your projects
This helps your science:
- Blogpost: Seven Reasons To Work Reproducibly (written by the Center of Open Science): https://cos.io/blog/seven-reasons-work-reproducibly/
- Have confidence in your analyses from this traceable, reusable record
- Save time through automation, thinking ahead of your immediate task, reduced bookkeeping, and collaboration
- Take advantage of convenient access: working openly online is like having an extended memory _ Making your data and code publicly available can increase the impact of research.
3. Learn these tools with collaborators and community (redefined):
- Your most important collaborator is Future You.
- Community should also be beyond the colleagues in your field.
- Learn from, with, and for others.
This helps your science:
- If you learn to talk about your data, you’ll find solutions faster.
- Build confidence: these skills are transferable beyond your science.
- Be empathetic and inclusive and build a network of allies
Build and/or join a local coding community
Join existing communities locally and online, and start local chapters with friends!
-
RLadies Dammam. Informal but efficient communities centered on R data analysis meant to be inclusive and supportive.
These meetups can be for skill-sharing, showcasing how people work, or building community so you can troubleshoot together. They can be an informal “hacky hour” at a cafe or pub!
Open Science Community Saudi Arabia
Open Science Community Saudi Arabia (OSCSA) was established in line with Saudi Arabia’s Vision 2030, which focuses on installing values, enhancing knowledge and improving equal access to education. It aims to provide a place where newcomers and experienced peers interact, inspire each other to embed open science practices and values in their workflows and provide feedback on policies, infrastructures, and support services. Our community is part of the International Network of Open Science & Scholarship Communities (INOSC).
Why Join the Community?
- Learn about Open Science practices through Workshops/Meetings.
- Support to lead your own project, and provide more visibility and discoverability for your work.
- OSCSA helps you to get connected with like-minded people from other communnities that adopt Open Science practices (e.g. Turing Way (UK), Carpentries, ArabR, …).
- Nominate yourself in the and vote for the international International Network of Open Science & Scholarship (INOSC) Board
Going further / Bibliography
- The Replication Crisis in Wikipedia
- A special issue in Science on Reproducibility
- Open Science And Reproducibility: presentation by Alexandra Sarafoglou (PhD student, UvA)
Key Points
Make your data and code available to others
Make your analyses reproducible
Make a sharp distincion between exploratory and confirmatory research
Introducing R and RStudio IDE
Overview
Teaching: 30 min
Exercises: 15 minQuestions
Why use R?
Why use RStudio and how does it differ from R?
Objectives
Know advantages of analyzing data in R
Know advantages of using RStudio
Create an RStudio project, and know the benefits of working within a project
Be able to customize the RStudio layout
Be able to locate and change the current working directory with
getwd()andsetwd()Compose an R script file containing comments and commands
Understand what an R function is
Locate help for an R function using
?,??, andargs()
Getting ready to use R for the first time
In this lesson we will take you through the very first things you need to get R working.
Tip: This lesson works best on the cloud
Remember, these lessons assume we are using the pre-configured virtual machine instances provided to you at a genomics workshop. Much of this work could be done on your laptop, but we use instances to simplify workshop setup requirements, and to get you familiar with using the cloud (a common requirement for working with big data). Visit the Genomics Workshop setup page for details on getting this instance running on your own, or for the info you need to do this on your own computer.
A Brief History of R
R has been around since 1995, and was created by Ross Ihaka and Robert Gentleman at the University of Auckland, New Zealand. R is based off the S programming language developed at Bell Labs and was developed to teach intro statistics. See this slide deck by Ross Ihaka for more info on the subject.
Advantages of using R
At more than 20 years old, R is fairly mature and growing in popularity. However, programming isn’t a popularity contest. Here are key advantages of analyzing data in R:
- R is open source. This means R is free - an advantage if you are at an institution where you have to pay for your own MATLAB or SAS license. Open source, is important to your colleagues in parts of the world where expensive software in inaccessible. It also means that R is actively developed by a community (see r-project.org), and there are regular updates.
- R is widely used. Ok, maybe programming is a popularity contest. Because, R is used in many areas (not just bioinformatics), you are more likely to find help online when you need it. Chances are, almost any error message you run into, someone else has already experienced.
- R is powerful. R runs on multiple platforms (Windows/MacOS/Linux). It can work with much larger datasets than popular spreadsheet programs like Microsoft Excel, and because of its scripting capabilities is far more reproducible. Also, there are thousands of available software packages for science, including genomics and other areas of life science.
Discussion: Your experience
What has motivated you to learn R? Have you had a research question for which spreadsheet programs such as Excel have proven difficult to use, or where the size of the data set created issues?
Introducing RStudio Server
In these lessons, we will be making use of a software called RStudio, an Integrated Development Environment (IDE). RStudio, like most IDEs, provides a graphical interface to R, making it more user-friendly, and providing dozens of useful features. We will introduce additional benefits of using RStudio as you cover the lessons. In this case, we are specifically using RStudio Server, a version of RStudio that can be accessed in your web browser. RStudio Server has the same features of the Desktop version of RStudio you could download as standalone software.
Log on to RStudio Cloud
Sign up on RStudio Cloud using either Google or GitHub.
Tip: If you have an account in GitHub, you are recommanded to use Github rather than Google.
You should now be looking at a page that will allow you to login to the RStudio cloud:
After signing up with GitHub, you should now see the RStudio Cloud:
Tip: Make sure there are no spaces before or after your URL or
your web browser may interpret it as a search query.
You should now be looking at a page that will allow you to login to the RStudio server:
Enter your user credentials and click Sign In. The credentials for the genomics Data Carpentry instances will be provided by your instructors.
You should now see the RStudio interface:
Create an RStudio project
One of the first benefits we will take advantage of in RStudio is something called an RStudio Project. An RStudio project allows you to more easily:
- Save data, files, variables, packages, etc. related to a specific analysis project
- Restart work where you left off
- Collaborate, especially if you are using version control such as git.
- To create a project, go to the File menu, and click New Project....
-
In the window that opens select New Directory, then New Project. For “Directory name:” enter dc_genomics_r. For “Create project as subdirectory of”, click Browse... and then click Choose which will select your home directory “~”.
-
Finally click Create Project. In the “Files” tab of your output pane (more about the RStudio layout in a moment), you should see an RStudio project file, dc_genomics_r.Rproj. All RStudio projects end with the “.Rproj” file extension.
Tip: Make your project more reproducible with renv
One of the most wonderful and also frustrating aspects of working with R is managing packages. We will talk more about them, but packages (e.g. ggplot2) are add-ons that extend what you can do with R. Unfortunately it is very common that you may run into versions of R and/or R packages that are not compatible. This may make it difficult for someone to run your R script using their version of R or a given R package, and/or make it more difficult to run their scripts on your machine. renv is an RStudio add-on that will associate your packages and project so that your work is more portable and reproducible. To turn on renv click on the Tools menu and select Project Options. Under Enviornments check off “Use renv with this project” and follow any installation instructions.
Creating your first R script
Now that we are ready to start exploring R, we will want to keep a record of the commands we are using. To do this we can create an R script:
Click the File menu and select New File and then R Script. Before we go any further, save your script by clicking the save/disk icon that is in the bar above the first line in the script editor, or click the File menu and select save. In the “Save File” window that opens, name your file “genomics_r_basics”. The new script genomics_r_basics.R should appear under “files” in the output pane. By convention, R scripts end with the file extension .R.
Overview and customization of the RStudio layout
Here are the major windows (or panes) of the RStudio environment:
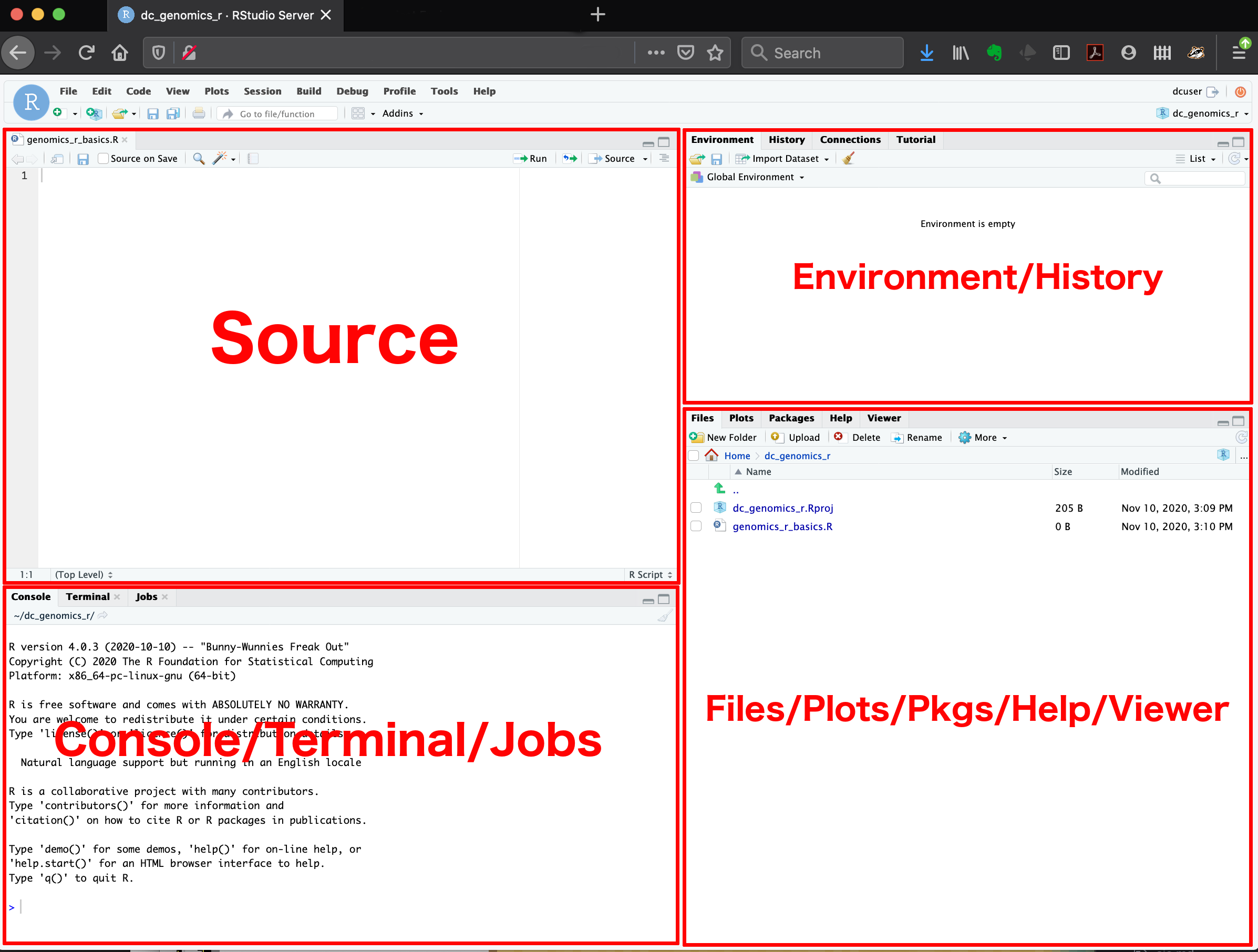
- Source: This pane is where you will write/view R scripts. Some outputs
(such as if you view a dataset using
View()) will appear as a tab here. - Console/Terminal/Jobs: This is actually where you see the execution of commands. This is the same display you would see if you were using R at the command line without RStudio. You can work interactively (i.e. enter R commands here), but for the most part we will run a script (or lines in a script) in the source pane and watch their execution and output here. The “Terminal” tab give you access to the BASH terminal (the Linux operating system, unrelated to R). RStudio also allows you to run jobs (analyses) in the background. This is useful if some analysis will take a while to run. You can see the status of those jobs in the background.
- Environment/History: Here, RStudio will show you what datasets and objects (variables) you have created and which are defined in memory. You can also see some properties of objects/datasets such as their type and dimensions. The “History” tab contains a history of the R commands you’ve executed R.
- Files/Plots/Packages/Help/Viewer: This multipurpose pane will show you the contents of directories on your computer. You can also use the “Files” tab to navigate and set the working directory. The “Plots” tab will show the output of any plots generated. In “Packages” you will see what packages are actively loaded, or you can attach installed packages. “Help” will display help files for R functions and packages. “Viewer” will allow you to view local web content (e.g. HTML outputs).
Tip: Uploads and downloads in the cloud
In the “Files” tab you can select a file and download it from your cloud instance (click the “more” button) to your local computer. Uploads are also possible.
All of the panes in RStudio have configuration options. For example, you can minimize/maximize a pane, or by moving your mouse in the space between panes you can resize as needed. The most important customization options for pane layout are in the View menu. Other options such as font sizes, colors/themes, and more are in the Tools menu under Global Options.
You are working with R
Although we won’t be working with R at the terminal, there are lots of reasons to. For example, once you have written an RScript, you can run it at any Linux or Windows terminal without the need to start up RStudio. We don’t want you to get confused - RStudio runs R, but R is not RStudio. For more on running an R Script at the terminal see this Software Carpentry lesson.
Getting to work with R: navigating directories
Now that we have covered the more aesthetic aspects of RStudio, we can get to work using some commands. We will write, execute, and save the commands we learn in our genomics_r_basics.R script that is loaded in the Source pane. First, lets see what directory we are in. To do so, type the following command into the script:
getwd()
To execute this command, make sure your cursor is on the same line the command is written. Then click the Run button that is just above the first line of your script in the header of the Source pane.
In the console, we expect to see the following output*:
[1] "/home/dcuser/dc_genomics_r"
* Notice, at the Console, you will also see the instruction you executed above the output in blue.
Since we will be learning several commands, we may already want to keep some
short notes in our script to explain the purpose of the command. Entering a #
before any line in an R script turns that line into a comment, which R will
not try to interpret as code. Edit your script to include a comment on the
purpose of commands you are learning, e.g.:
# this command shows the current working directory
getwd()
Exercise: Work interactively in R
What happens when you try to enter the
getwd()command in the Console pane?Solution
You will get the same output you did as when you ran
getwd()from the source. You can run any command in the Console, however, executing it from the source script will make it easier for us to record what we have done, and ultimately run an entire script, instead of entering commands one-by-one.
For the purposes of this exercise we want you to be in the directory "/home/dcuser/R_data".
What if you weren’t? You can set your home directory using the setwd()
command. Enter this command in your script, but don’t run this yet.
# This sets the working directory
setwd()
You may have guessed, you need to tell the setwd() command
what directory you want to set as your working directory. To do so, inside of
the parentheses, open a set of quotes. Inside the quotes enter a / which is
the root directory for Linux. Next, use the Tab key, to take
advantage of RStudio’s Tab-autocompletion method, to select home, dcuser,
and dc_genomics_r directory. The path in your script should look like this:
# This sets the working directory
setwd("/home/dcuser/dc_genomics_r")
When you run this command, the console repeats the command, but gives you no
output. Instead, you see the blank R prompt: >. Congratulations! Although it
seems small, knowing what your working directory is and being able to set your
working directory is the first step to analyzing your data.
Tip: Never use
setwd()Wait, what was the last 2 minutes about? Well, setting your working directory is something you need to do, you need to be very careful about using this as a step in your script. For example, what if your script is being on a computer that has a different directory structure? The top-level path in a Unix file system is root
/, but on Windows it is likelyC:\. This is one of several ways you might cause a script to break because a file path is configured differently than your script anticipates. R packages like here and file.path allow you to specify file paths is a way that is more operating system independent. See Jenny Bryan’s blog post for this and other R tips.
Using functions in R, without needing to master them
A function in R (or any computing language) is a short
program that takes some input and returns some output. Functions may seem like an advanced topic (and they are), but you have already
used at least one function in R. getwd() is a function! The next sections will help you understand what is happening in
any R script.
Exercise: What do these functions do?
Try the following functions by writing them in your script. See if you can guess what they do, and make sure to add comments to your script about your assumed purpose.
dir()sessionInfo()date()Sys.time()Solution
dir()# Lists files in the working directorysessionInfo()# Gives the version of R and additional info including on attached packagesdate()# Gives the current dateSys.time()# Gives the current timeNotice: Commands are case sensitive!
You have hopefully noticed a pattern - an R function has three key properties:
- Functions have a name (e.g.
dir,getwd); note that functions are case sensitive! - Following the name, functions have a pair of
() - Inside the parentheses, a function may take 0 or more arguments
An argument may be a specific input for your function and/or may modify the
function’s behavior. For example the function round() will round a number
with a decimal:
# This will round a number to the nearest integer
round(3.14)
[1] 3
Getting help with function arguments
What if you wanted to round to one significant digit? round() can
do this, but you may first need to read the help to find out how. To see the help
(In R sometimes also called a “vignette”) enter a ? in front of the function
name:
?round()
The “Help” tab will show you information (often, too much information). You
will slowly learn how to read and make sense of help files. Checking the “Usage” or “Examples”
headings is often a good place to look first. If you look under “Arguments,” we
also see what arguments we can pass to this function to modify its behavior.
You can also see a function’s argument using the args() function:
args(round)
function (x, digits = 0)
NULL
round() takes two arguments, x, which is the number to be
rounded, and a
digits argument. The = sign indicates that a default (in this case 0) is
already set. Since x is not set, round() requires we provide it, in contrast
to digits where R will use the default value 0 unless you explicitly provide
a different value. We can explicitly set the digits parameter when we call the
function:
round(3.14159, digits = 2)
[1] 3.14
Or, R accepts what we call “positional arguments”, if you pass a function
arguments separated by commas, R assumes that they are in the order you saw
when we used args(). In the case below that means that x is 3.14159 and
digits is 2.
round(3.14159, 2)
[1] 3.14
Finally, what if you are using ? to get help for a function in a package not installed on your system, such as when you are running a script which has dependencies.
?geom_point()
will return an error:
Error in .helpForCall(topicExpr, parent.frame()) :
no methods for ‘geom_point’ and no documentation for it as a function
Use two question marks (i.e. ??geom_point()) and R will return
results from a search of the documentation for packages you have installed on your computer
in the “Help” tab. Finally, if you think there
should be a function, for example a statistical test, but you aren’t
sure what it is called in R, or what functions may be available, use
the help.search() function.
Exercise: Searching for R functions
Use
help.search()to find R functions for the following statistical functions. Remember to put your search query in quotes inside the function’s parentheses.
- Chi-Squared test
- Student t-test
- mixed linear model
Solution
While your search results may return several tests, we list a few you might find:
- Chi-Squared test:
stats::Chisquare- Student t-test:
stats::t.test- mixed linear model:
stats::lm.glm
We will discuss more on where to look for the libraries and packages that contain functions you want to use. For now, be aware that two important ones are CRAN - the main repository for R, and Bioconductor - a popular repository for bioinformatics-related R packages.
RStudio contextual help
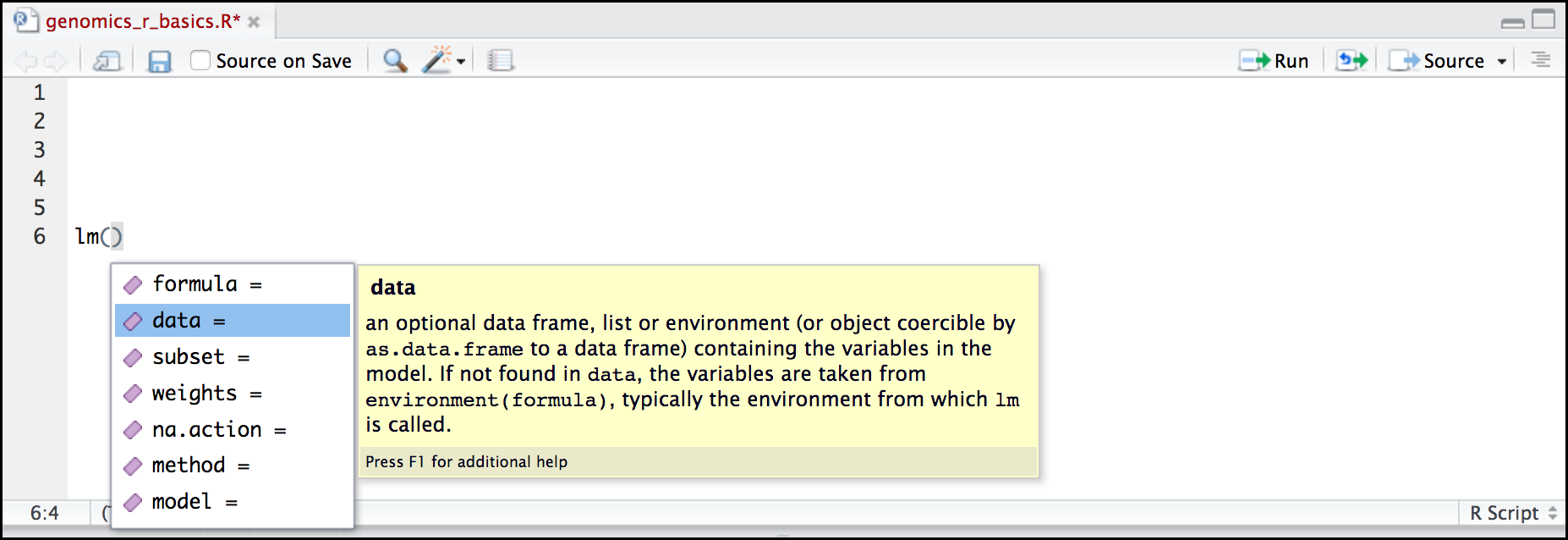
Here is one last bonus we will mention about RStudio. It’s difficult to remember all of the arguments and definitions associated with a given function. When you start typing the name of a function and hit the Tab key, RStudio will display functions and associated help:
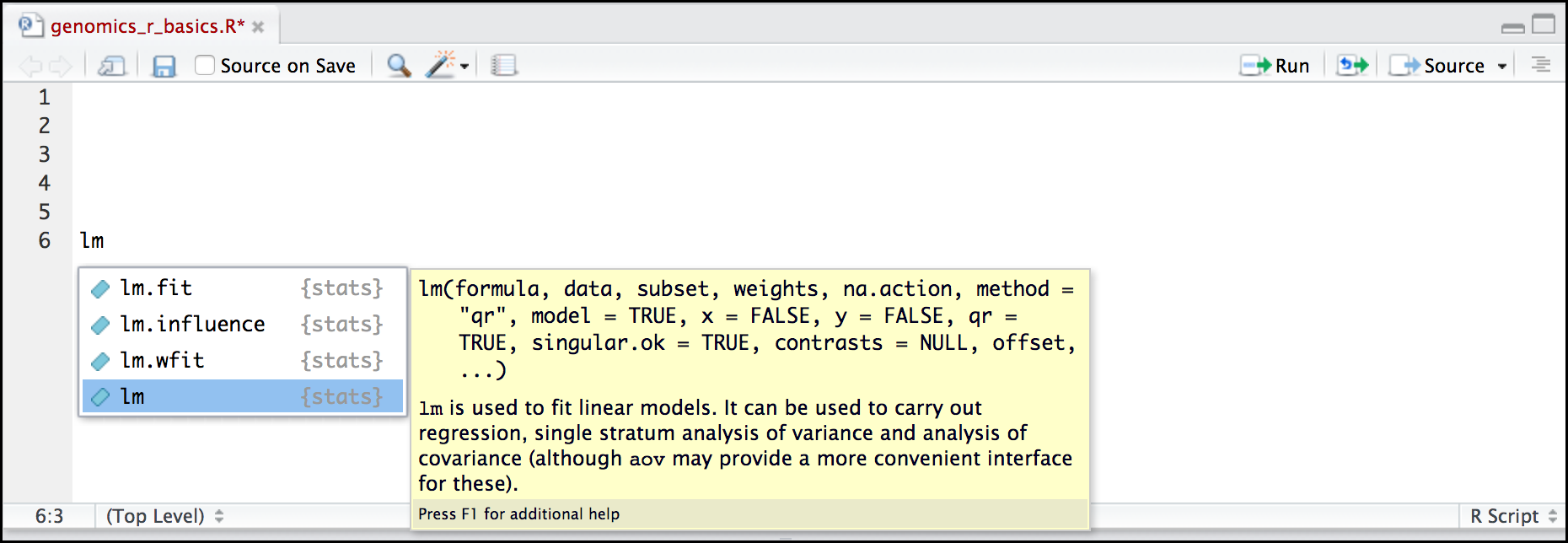
Once you type a function, hitting the Tab inside the parentheses will show you the function’s arguments and provide additional help for each of these arguments.
Key Points
R is a powerful, popular open-source scripting language
You can customize the layout of RStudio, and use the project feature to manage the files and packages used in your analysis
RStudio allows you to run R in an easy-to-use interface and makes it easy to find help
Collaborating with Github
Overview
Teaching: 30 min
Exercises: 60 minQuestions
How can I develop and collaborate on code with another scientist?
How can I give access to my code to another collaborator?
How can I keep code synchronised with another scientist?
How can I solve conflicts that arise from that collaboration?
What are Github
Objectives
Be able to create a new repository and share it with another scientist.
Be able to work together on a R script through RStudio and Github integration.
Understand how to make issues and explore the history of a repository.
Table of contents
Introduction
The collaborative power of GitHub and RStudio is really game changing. So far we’ve been collaborating with our most important collaborator: ourselves. But, we are lucky that in science we have so many other collaborators, so let’s learn how to accelerate our collaborations with them through GitHub!
We are going to teach you the simplest way to collaborate with someone, which is for both of you to have privileges to edit and add files to a repository. GitHub is built for software developer teams but we believe that it can also be beneficial to scientists.
We will do this all with a partner, and we’ll walk through some things all together, and then give you a chance to work with your collaborator on your own.
Pair up and work collaboratively
- Make groups of two scientists. They will collaborate through Github.
- Decide who will own the Github repository: this will be the “owner” also referred to as Partner 1.
- The other scientist will be called the “collaborator” also referred to as Partner 2.
- Please write your role on a sticky note and place it on your laptop to remember who you are!
Owner (Partner 1) setup
Create a Github repository
The repository “owner” will connect to Github and create a repository called first-collaboration. We will do this in the same way that we did in the “Version control with git and Github” episode.
Create a gh-pages branch
We aren’t going to talk about branches very much, but they are a powerful feature of git and GitHub. I think of it as creating a copy of your work that becomes a parallel universe that you can modify safely because it’s not affecting your original work. And then you can choose to merge the universes back together if and when you want.
By default, when you create a new repo you begin with one branch, and it is named master. When you create new branches, you can name them whatever you want. However, if you name one gh-pages (all lowercase, with a - and no spaces), this will let you create a website. And that’s our plan. So, owner/partner 1, please do this to create a gh-pages branch:
On the homepage for your repo on GitHub.com, click the button that says “Branch:master”. Here, you can switch to another branch (right now there aren’t any others besides master), or create one by typing a new name.
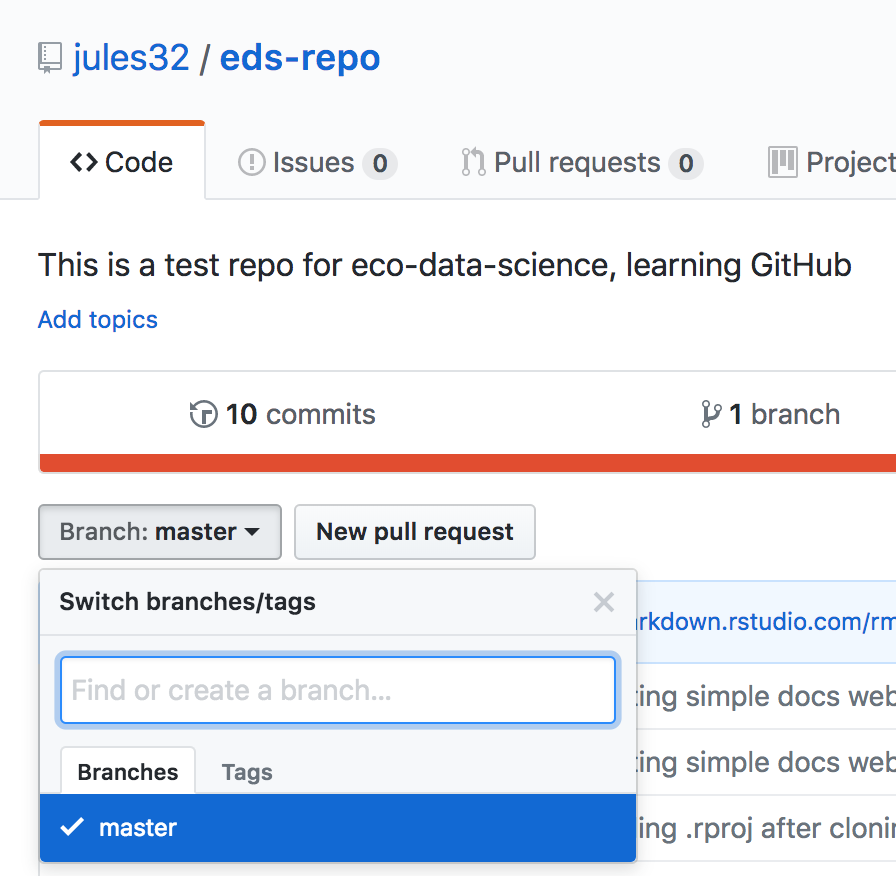
Let’s type gh-pages.
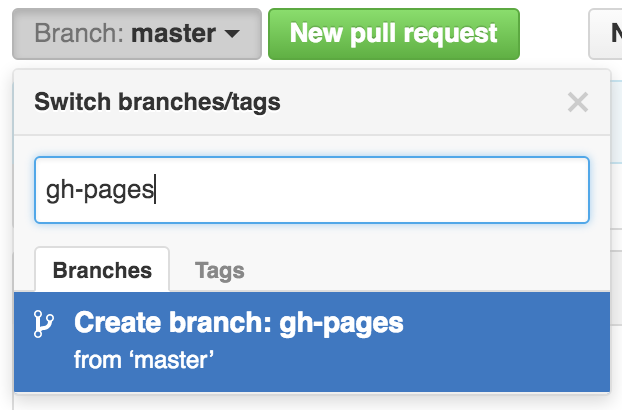
Let’s also change gh-pages to the default branch and delete the master branch: this will be a one-time-only thing that we do here:
First click to control branches:
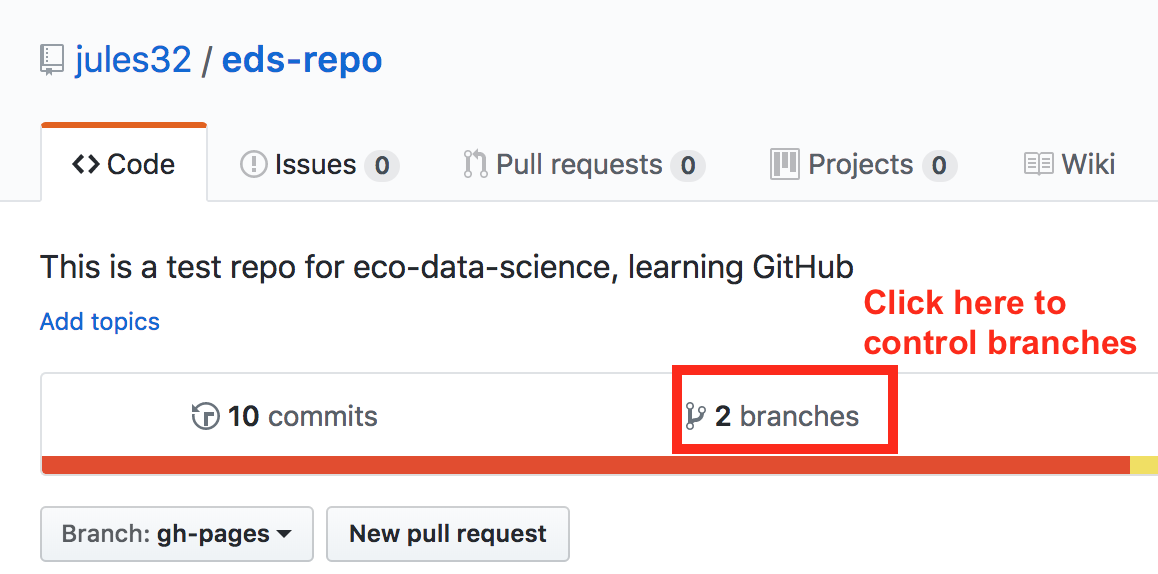
And then click to change the default branch to gh-pages. I like to then delete the master branch when it has the little red trash can next to it. It will make you confirm that you really want to delete it, which I do!
Give your collaborator administration privileges (Partner 1 and 2)
Now, Partner 1, go into Settings > Collaborators > enter Partner 2’s (your collaborator’s) username.
Partner 2 then needs to check their email and accept as a collaborator. Notice that your collaborator has “Push access to the repository” (highlighted below):

Clone to a new Rproject (Owner Partner 1)
Now let’s have Partner 1 clone the repository to their local computer. We’ll do this through RStudio like we did before (see the “Version control with git and Github:Clone your repository using RStudio” episode section. But, we’ll do this with a final additional step before hitting the “Create Project”: we will select “Open in a new Session”.

Opening this Project in a new Session opens up a new world of awesomeness from RStudio. Having different RStudio project sessions allows you to keep your work separate and organized. So you can collaborate with this collaborator on this repository while also working on your other repository from this morning. I tend to have a lot of projects going at one time:
Have a look in your git tab.
Like we saw earlier, when you first clone a repo through RStudio, RStudio will add an .Rproj file to your repo. And if you didn’t add a .gitignore file when you originally created the repo on GitHub.com, RStudio will also add this for you. So, Partner 1, let’s go ahead and sync this back to GitHub.com.
Remember:
Let’s confirm that this was synced by looking at GitHub.com again. You may have to refresh the page, but you should see this commit where you added the .Rproj file.
Collaborator (Partner 2) part
Clone to a new Rproject (Partner 2)
Now it’s Partner 2’s turn! Partner 2, clone this repository following the same steps that Partner 1 just did. When you clone it, RStudio should not create any new files — why? Partner 1 already created and pushed the .Rproj and .gitignore files so they already exist in the repo.
Discussion point
Question: When you clone it, RStudio should not create any new files — why?
Solution
Partner 1 already created and pushed the
.Rprojand.gitignorefiles so they already exist in the repo.
Edit a file and sync (Partner 2)
Let’s have Partner 2 add some information to the README.md. Let’s have them write:
Collaborators:
- Partner 2's name
When we save the README.md, And now let’s sync back to GitHub.
When we inspect on GitHub.com, click to view all the commits, you’ll see commits logged from both Partner 1 and 2!
Discussion point
Questions:
- Would you be able to clone a repository that you are not a collaborator on?
- What do you think would happen? Try it!
- Can you sync back?
Solution
- Yes, you can clone a repository that is publicly available.
- If you try to clone it on your local machine, it does work.
- Unfortunately, if you don’t have write permissions, you cannot contribute. You would have to ask for write/push writes.
State of the Repository
OK, so where do things stand right now? GitHub.com has the most recent versions of all the repository’s files. Partner 2 also has these most recent versions locally. How about Partner 1?
Partner 1 does not have the most recent versions of everything on their computer!.
Discussion point
Question: How can we change that? Or how could we even check?
Solution
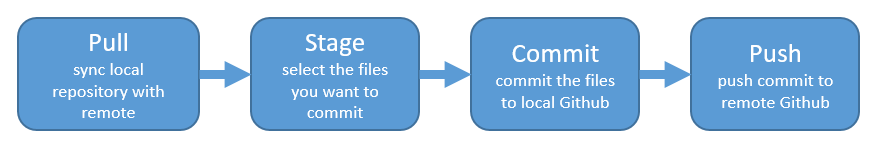
PULL !
Let’s have Partner 1 go back to RStudio and Pull. If their files aren’t up-to-date, this will pull the most recent versions to their local computer. And if they already did have the most recent versions? Well, pulling doesn’t cost anything (other than an internet connection), so if everything is up-to-date, pulling is fine too.
I recommend pulling every time you come back to a collaborative repository. Whether you haven’t opened RStudio in a month or you’ve just been away for a lunch break, pull. It might not be necessary, but it can save a lot of heartache later.
Merge conflicts
What kind of heartache are we talking about? Let’s explore.
Stop and watch me create and solve a merge conflict with my Partner 2, and then you will have time to recreate this with your partner.
Within a file, GitHub tracks changes line-by-line. So you can also have collaborators working on different lines within the same file and GitHub will be able to weave those changes into each other – that’s it’s job! It’s when you have collaborators working on the same lines within the same file that you can have merge conflicts. Merge conflicts can be frustrating, but they are actually trying to help you (kind of like R’s error messages). They occur when GitHub can’t make a decision about what should be on a particular line and needs a human (you) to decide. And this is good – you don’t want GitHub to decide for you, it’s important that you make that decision.
Me = partner 1. My co-instructor = partner 2.
Here’s what me and my collaborator are going to do:
- My collaborator and me are first going to pull.
- Then, my collaborator and me navigate to the README file within RStudio.
- My collaborator and me are going to write something in the same file on the same line. We are going to write something in the README file on line 7: for instance, “I prefer R” and “I prefer Python”.
- Save the README file.
- My collaborator is going to pull, stage, commit and push.
- When my collaborator is done, I am going to pull.
- Error! Merge conflict!

I am not allowed to to pull since GitHub is protecting me because if I did successfully pull, my work would be overwritten by whatever my collaborator had written.
GitHub is going to make a human (me in this case) decide. GitHub says, “either commit this work first, or stash it”. Stashing means “ (“save a copy of the README in another folder somewhere outside of this GitHub repository”).
Let’s follow their advice and have me to commit first. Great. Now let’s pull again.
Still not happy!
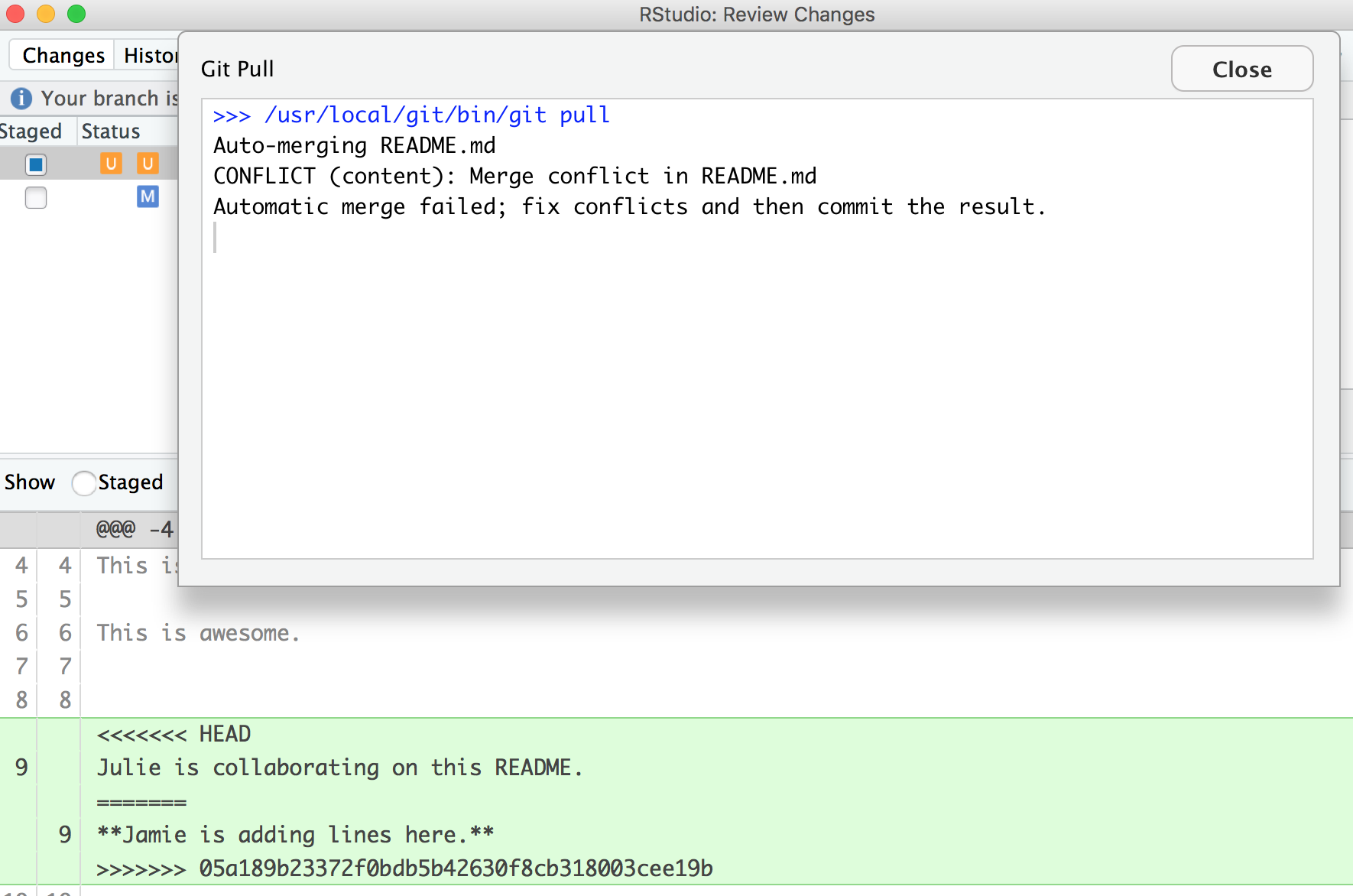
OK, actually, we’re just moving along this same problem that we know that we’ve created: Both me and my collaborator have both added new information to the same line. You can see that the pop-up box is saying that there is a CONFLICT and the merge has not happened. OK. We can close that window and inspect.
Notice that in the git tab, there are orange Us; this means that there is an unresolved conflict, and it is not staged with a check anymore because modifications have occurred to the file since it has been staged.
Let’s look at the README file itself. We got a preview in the diff pane that there is some new text going on in our README file:
<<<<<<< HEAD
Julie is collaborating on this README.
=======
**Jamie is adding lines here.**
>>>>>>> 05a189b23372f0bdb5b42630f8cb318003cee19b
In this example, I am Jamie and my collaborator is Julie. GitHub is displaying the line that Julie wrote and the line Jamie wrote separated by =======. So these are the two choices that Partner 2 has to decide between, which one do you want to keep? Where where does this decision start and end? The lines are bounded by <<<<<<<HEAD and >>>>>>>long commit identifier.
So, to resolve this merge conflict, my collaborator has to chose, and delete everything except the line they want. So, they will delete the <<<<<<HEAD, =====, >>>>long commit identifier and one of the lines that they don’t want to keep.
Do that, and let’s try again. In this example, we’ve kept my (Jamie’s) line:
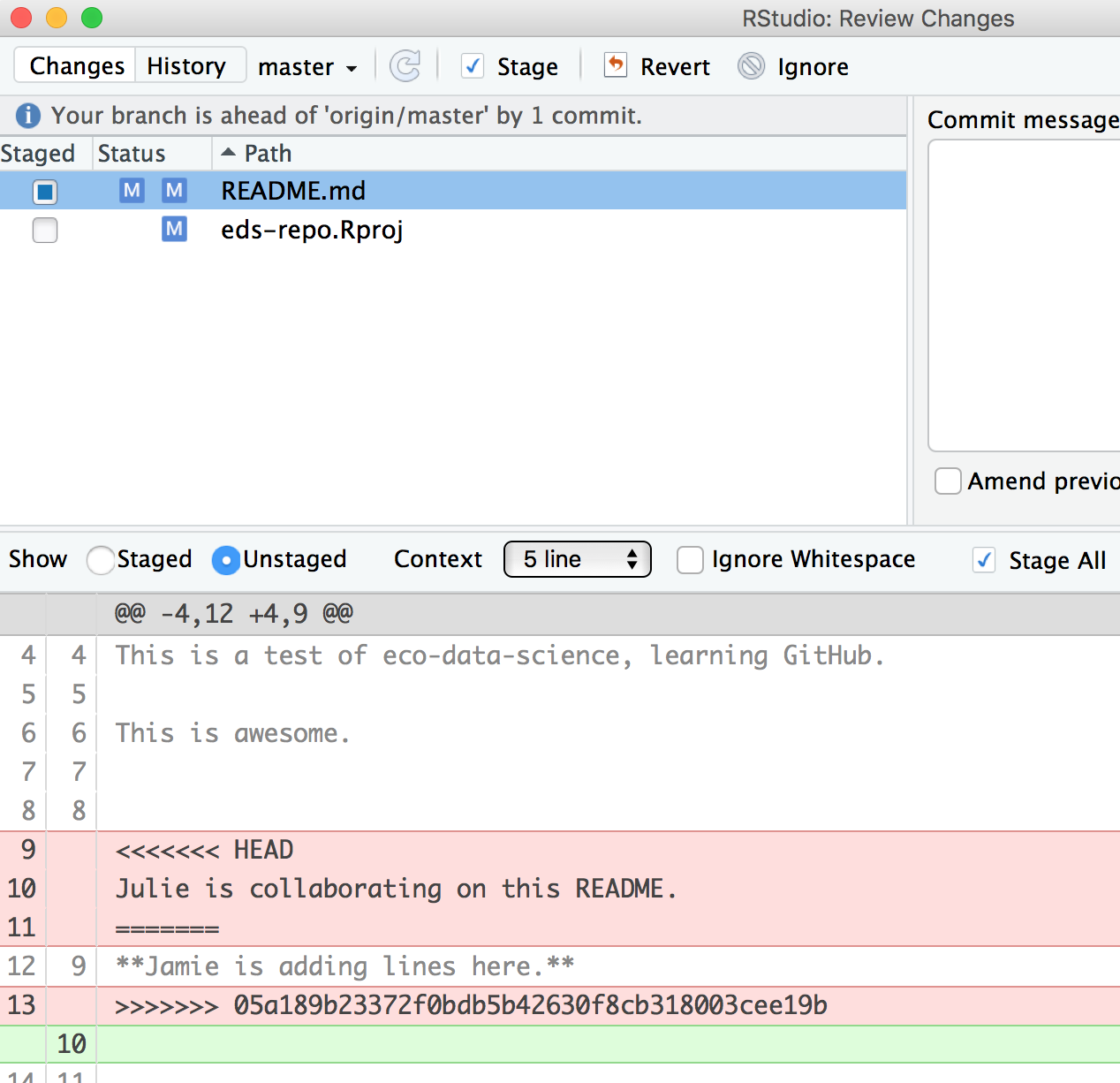
Then be I need to stage and write a commit message. I often write “resolving merge conflict” or something so I know what I was up to. When I stage the file, notice how now my edits look like a simple line replacement (compare with the image above before it was re-staged):

Your turn
Exercise
- Create a merge conflict with your partner, like we did in the example above.
- Try to fix it.
- Try other ways to get and solve merge conflicts. For example, when you get the following error message, try both ways (commit or stash. Stash means copy/move it somewhere else, for example, on your Desktop temporarily).
How do you avoid merge conflicts?
I’d say pull often, commit and sync often.
Also, talk with your collaborators. Even on a very collaborative project (e.g. a scientific publication), you are actually rarely working on the exact same file at any given time. And if you are, make sure you talk in-person or through chat applications (Slack, Gitter, Whatsapp, etc.).
But merge conflicts will occur and some of them will be heartbreaking and demoralizing. They happen to me when I collaborate with myself between my work computer and laptop. So protect yourself by pulling and syncing often!
Create your collaborative website
OK. Let’s have Partner 2 create a new RMarkdown file. Here’s what they will do:
- Pull!
- Create a new RMarkdown file and name it
index.Rmd. Make sure it’s all lowercase, and namedindex.Rmd. This will be the homepage for our website! - Maybe change the title inside the Rmd, call it “Our website”
- Knit!
- Save and sync your .Rmd and your .html files: pull, stage, commit, pull, push.
- Go to GitHub.com and go to your rendered website! Where is it? Figure out your website’s url from your github repo’s url. For example:
- my github repo: https://github.com/jules32/collab-research
- my website url: https://jules32.github.io/collab-research/
- note that the url starts with my username.github.io
So cool! On websites, if something is called index.html, that defaults to the home page. So https://jules32.github.io/collab-research/ is the same as https://jules32.github.io/collab-research/index.html. If you name your RMarkdown file my_research.Rmd, the url will become https://jules32.github.io/collab-research/my_research.html.
Your turn
Exercise
Here is some collaborative analysis you can do on your own. We’ll be playing around > with airline flights data, so let’s get setup a bit.
- Person 1: clean up the README to say something about you two, the authors.
- Person 2: edit the
index.Rmdor create a new RMarkdown file: maybe add something about the authors, and knit it.- Both of you: sync to GitHub.com (pull, stage, commit, push).
- Both of you: once you’ve both synced (talk to each other about it!), pull again. You should see each others’ work on your computer.
- Person 1: in the RMarkdown file, add a bit of the plan. We’ll be exploring the
nycflights13dataset. This is data on flights departing New York City in 2013.- Person 2: in the README, add a bit of the plan.
- Both of you: sync
Explore on GitHub.com
Now, let’s look at the repo again on GitHub.com. You’ll see those new files appear, and the commit history has increased.
Commit History
You’ll see that the number of commits for the repo has increased, let’s have a look. You can see the history of both of you.
Blame
Now let’s look at a single file, starting with the README file. We’ve explored the “Raw” and “History” options in the top-right of the file, but we haven’t really explored the “Blame” option. Let’s look now. Blame shows you line-by-line who authored the most recent version of the file you see. This is super useful if you’re trying to understand logic; you know who to ask for questions or attribute credit.
Issues
Now let’s have a look at issues. This is a way you can communicate to others about plans for the repo, questions, etc. Note that issues are public if the repository is public.

Let’s create a new issue with the title “NYC flights”.
In the text box, let’s write a note to our collaborator. You can use the Markdown syntax in this text box, which means all of your header and bullet formatting will come through. You can also select these options by clicking them just above the text box.
Let’s have one of you write something here. I’m going to write:
Hi @jafflerbach!
# first priority
- explore NYC flights
- plot interesting things
Note that I have my collaborator’s GitHub name with a @ symbol. This is going to email her directly so that she sees this issue. I can click the “Preview” button at the top left of the text box to see how this will look rendered in Markdown. It looks good!
Now let’s click submit new issue.
On the right side, there are a bunch of options for categorizing and organizing your issues. You and your collaborator may want to make some labels and timelines, depending on the project.
Another feature about issues is whether you want any notifications to this repository. Click where it says “Unwatch” up at the top. You’ll see three options: “Not watching”, “Watching”, and “Ignoring”. By default, you are watching these issues because you are a collaborator to the repository. But if you stop being a big contributor to this project, you may want to switch to “Not watching”. Or, you may want to ask an outside person to watch the issues. Or you may want to watch another repo yourself!
Let’s have Person 2 respond to the issue affirming the plan.
NYC flights exploration
Let’s continue this workflow with your collaborator, syncing to GitHub often and practicing what we’ve learned so far. We will get started together and then you and your collaborator will work on your own.
Here’s what we’ll be doing (from R for Data Science’s Transform Chapter):
Data: You will be exploring a dataset on flights departing New York City in 2013. These data are actually in a package called nycflights13, so we can load them the way we would any other package.
Let’s have Person 1 write this in the RMarkdown document (Partner 2 just listen for a moment; we will sync this to you in a moment).
library(nycflights13) # install.packages('nycflights13')
library(tidyverse)
This data frame contains all flights that departed from New York City in 2013. The data comes from the US Bureau of Transportation Statistics, and is documented in ?flights.
flights
Let’s select all flights on January 1st with:
filter(flights, month == 1, day == 1)
To use filtering effectively, you have to know how to select the observations that you want using the comparison operators. R provides the standard suite: >, >=, <, <=, != (not equal), and == (equal). We learned these operations yesterday. But there are a few others to learn as well.
Sync
Sync this RMarkdown back to GitHub so that your collaborator has access to all these notes.
Partner 2 pull
Now is the time to pull!
Partner 2 will continue with the following notes and instructions:
Logical operators
Multiple arguments to filter() are combined with “and”: every expression must be true in order for a row to be included in the output. For other types of combinations, you’ll need to use Boolean operators yourself:
&is “and”|is “or”!is “not”
Let’s have a look:
The following code finds all flights that departed in November or December:
filter(flights, month == 11 | month == 12)
The order of operations doesn’t work like English. You can’t write filter(flights, month == 11 | 12), which you might literally translate into “finds all flights that departed in November or December”. Instead it finds all months that equal 11 | 12, an expression that evaluates to TRUE. In a numeric context (like here), TRUE becomes one, so this finds all flights in January, not November or December. This is quite confusing!
A useful short-hand for this problem is x %in% y. This will select every row where x is one of the values in y. We could use it to rewrite the code above:
nov_dec <- filter(flights, month %in% c(11, 12))
Sometimes you can simplify complicated subsetting by remembering De Morgan’s law: !(x & y) is the same as !x | !y, and !(x | y) is the same as !x & !y. For example, if you wanted to find flights that weren’t delayed (on arrival or departure) by more than two hours, you could use either of the following two filters:
filter(flights, !(arr_delay > 120 | dep_delay > 120))
filter(flights, arr_delay <= 120, dep_delay <= 120)
Whenever you start using complicated, multipart expressions in filter(), consider making them explicit variables instead. That makes it much easier to check your work.
Partner 2 sync
Once you have filtered the flights dataframe for flights, sync it to Github (add, commit and push).
Your turn
Based on what you’ve learned previously about data transformation, you’ll make a series of data transformation on the flights dataset. Some ideas:
- calculate the average flight delay.
- determine the longest flight distance.
- Your own question!
Exercise
Partner 1 will pull so that we all have the most current information. With your partner, transform and compute several metrics about the data. Partner 1 and 2, make sure you talk to each other and decide on who does what. Remember to make your commit messages useful! As you work, you may get merge conflicts. This is part of collaborating in GitHub; we will walk through and help you with these and also teach the whole group.
Key Points
Github allows you to synchronise work efforts and collaborate with other scientists on (R) code.
Github can be used to make custom website visible on the internet.
Merge conflicts can arise between you and yourself (different machines).
Merge conflicts arise when you collaborate and are a safe way to handle discordance.
Efficient collaboration on data analysis can be made using Github.
R Basics
Overview
Teaching: 60 min
Exercises: 20 minQuestions
What will these lessons not cover?
What are the basic features of the R language?
What are the most common objects in R?
Objectives
Be able to create the most common R objects including vectors
Understand that vectors have modes, which correspond to the type of data they contain
Be able to use arithmetic operators on R objects
Be able to retrieve (subset), name, or replace, values from a vector
Be able to use logical operators in a subsetting operation
Understand that lists can hold data of more than one mode and can be indexed
“The fantastic world of R awaits you” OR “Nobody wants to learn how to use R”
Before we begin this lesson, we want you to be clear on the goal of the workshop and these lessons. We believe that every learner can achieve competency with R. You have reached competency when you find that you are able to use R to handle common analysis challenges in a reasonable amount of time (which includes time needed to look at learning materials, search for answers online, and ask colleagues for help). As you spend more time using R (there is no substitute for regular use and practice) you will find yourself gaining competency and even expertise. The more familiar you get, the more complex the analyses you will be able to carry out, with less frustration, and in less time - the fantastic world of R awaits you!
What these lessons will not teach you
Nobody wants to learn how to use R. People want to learn how to use R to analyze their own research questions! Ok, maybe some folks learn R for R’s sake, but these lessons assume that you want to start analyzing genomic data as soon as possible. Given this, there are many valuable pieces of information about R that we simply won’t have time to cover. Hopefully, we will clear the hurdle of giving you just enough knowledge to be dangerous, which can be a high bar in R! We suggest you look into the additional learning materials in the tip box below.
Here are some R skills we will not cover in these lessons
- How to create and work with R matrices and R lists
- How to create and work with loops and conditional statements, and the “apply” family of functions (which are super useful, read more here)
- How to do basic string manipulations (e.g. finding patterns in text using grep, replacing text)
- How to plot using the default R graphic tools (we will cover plot creation, but will do so using the popular plotting package
ggplot2) - How to use advanced R statistical functions
Tip: Where to learn more
The following are good resources for learning more about R. Some of them can be quite technical, but if you are a regular R user you may ultimately need this technical knowledge.
- R for Beginners: By Emmanuel Paradis and a great starting point
- The R Manuals: Maintained by the R project
- R contributed documentation: Also linked to the R project; importantly there are materials available in several languages
- R for Data Science: A wonderful collection by noted R educators and developers Garrett Grolemund and Hadley Wickham
- Practical Data Science for Stats: Not exclusively about R usage, but a nice collection of pre-prints on data science and applications for R
- Programming in R Software Carpentry lesson: There are several Software Carpentry lessons in R to choose from
Creating objects in R
Reminder
At this point you should be coding along in the “genomics_r_basics.R” script we created in the last episode. Writing your commands in the script (and commenting it) will make it easier to record what you did and why.
What might be called a variable in many languages is called an object in R.
To create an object you need:
- a name (e.g. ‘a’)
- a value (e.g. ‘1’)
- the assignment operator (‘<-‘)
In your script, “genomics_r_basics.R”, using the R assignment operator ‘<-‘, assign ‘1’ to the object ‘a’ as shown. Remember to leave a comment in the line above (using the ‘#’) to explain what you are doing:
# this line creates the object 'a' and assigns it the value '1'
a <- 1
Next, run this line of code in your script. You can run a line of code by hitting the Run button that is just above the first line of your script in the header of the Source pane or you can use the appropriate shortcut:
- Windows execution shortcut: Ctrl+Enter
- Mac execution shortcut: Cmd(⌘)+Enter
To run multiple lines of code, you can highlight all the line you wish to run and then hit Run or use the shortcut key combo listed above.
In the RStudio ‘Console’ you should see:
a <- 1
>
The ‘Console’ will display lines of code run from a script and any outputs or status/warning/error messages (usually in red).
In the ‘Environment’ window you will also get a table:
| Values | |
|---|---|
| a | 1 |
The ‘Environment’ window allows you to keep track of the objects you have created in R.
Exercise: Create some objects in R
Create the following objects; give each object an appropriate name (your best guess at what name to use is fine):
- Create an object that has the value of number of pairs of human chromosomes
- Create an object that has a value of your favorite gene name
- Create an object that has this URL as its value: “ftp://ftp.ensemblgenomes.org/pub/bacteria/release-39/fasta/bacteria_5_collection/escherichia_coli_b_str_rel606/”
- Create an object that has the value of the number of chromosomes in a diploid human cell
Solution
Here as some possible answers to the challenge:
human_chr_number <- 23 gene_name <- 'pten' ensemble_url <- 'ftp://ftp.ensemblgenomes.org/pub/bacteria/release-39/fasta/bacteria_5_collection/escherichia_coli_b_str_rel606/' human_diploid_chr_num <- 2 * human_chr_number
Naming objects in R
Here are some important details about naming objects in R.
- Avoid spaces and special characters: Object names cannot contain spaces or the minus sign (
-). You can use ‘_’ to make names more readable. You should avoid using special characters in your object name (e.g. ! @ # . , etc.). Also, object names cannot begin with a number. - Use short, easy-to-understand names: You should avoid naming your objects using single letters (e.g. ‘n’, ‘p’, etc.). This is mostly to encourage you to use names that would make sense to anyone reading your code (a colleague, or even yourself a year from now). Also, avoiding excessively long names will make your code more readable.
- Avoid commonly used names: There are several names that may already have a definition in the R language (e.g. ‘mean’, ‘min’, ‘max’). One clue that a name already has meaning is that if you start typing a name in RStudio and it gets a colored highlight or RStudio gives you a suggested autocompletion you have chosen a name that has a reserved meaning.
- Use the recommended assignment operator: In R, we use ‘<- ‘ as the
preferred assignment operator. ‘=’ works too, but is most commonly used in
passing arguments to functions (more on functions later). There is a shortcut
for the R assignment operator:
- Windows execution shortcut: Alt+-
- Mac execution shortcut: Option+-
There are a few more suggestions about naming and style you may want to learn more about as you write more R code. There are several “style guides” that have advice, and one to start with is the tidyverse R style guide.
Tip: Pay attention to warnings in the script console
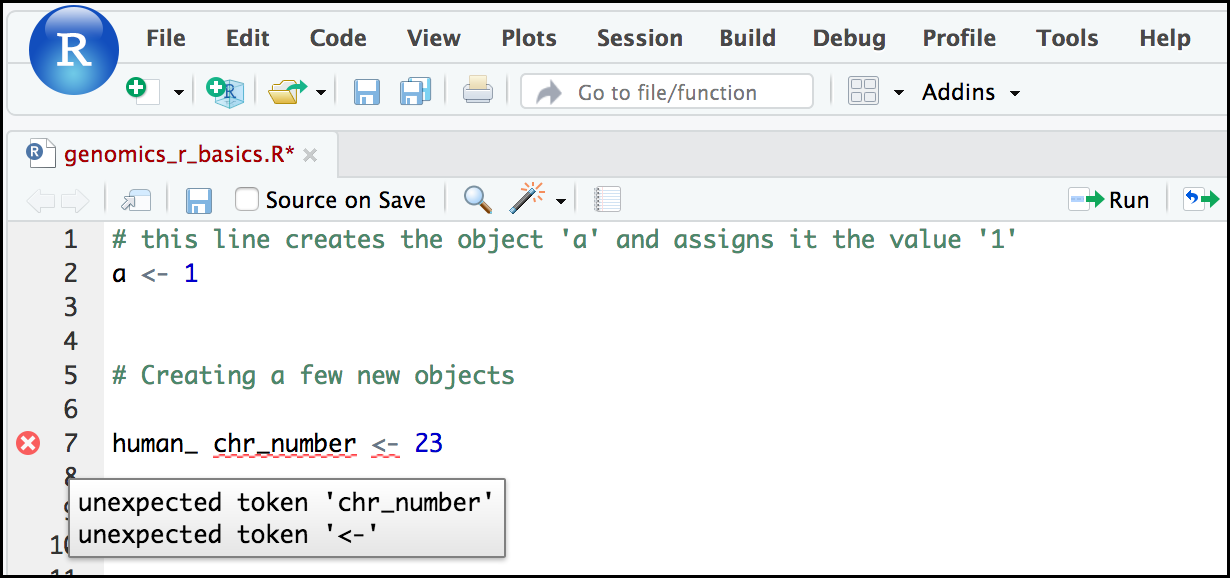
If you enter a line of code in your script that contains an error, RStudio may give you an error message and underline this mistake. Sometimes these messages are easy to understand, but often the messages may need some figuring out. Paying attention to these warnings will help you avoid mistakes. In the example below, our object name has a space, which is not allowed in R. The error message does not say this directly, but R is “not sure” about how to assign the name to “human_ chr_number” when the object name we want is “human_chr_number”.
Reassigning object names or deleting objects
Once an object has a value, you can change that value by overwriting it. R will not give you a warning or error if you overwriting an object, which may or may not be a good thing depending on how you look at it.
# gene_name has the value 'pten' or whatever value you used in the challenge.
# We will now assign the new value 'tp53'
gene_name <- 'tp53'
You can also remove an object from R’s memory entirely. The rm() function
will delete the object.
# delete the object 'gene_name'
rm(gene_name)
If you run a line of code that has only an object name, R will normally display the contents of that object. In this case, we are told the object no longer exists.
Error: object 'gene_name' not found
Understanding object data types (modes)
In R, every object has two properties:
- Length: How many distinct values are held in that object
- Mode: What is the classification (type) of that object.
We will get to the “length” property later in the lesson. The “mode” property corresponds to the type of data an object represents. The most common modes you will encounter in R are:
| Mode (abbreviation) | Type of data |
|---|---|
| Numeric (num) | Numbers such floating point/decimals (1.0, 0.5, 3.14), there are also more specific numeric types (dbl - Double, int - Integer). These differences are not relevant for most beginners and pertain to how these values are stored in memory |
| Character (chr) | A sequence of letters/numbers in single ‘’ or double “ “ quotes |
| Logical | Boolean values - TRUE or FALSE |
There are a few other modes (i.e. “complex”, “raw” etc.) but these are the three we will work with in this lesson.
Data types are familiar in many programming languages, but also in natural language where we refer to them as the parts of speech, e.g. nouns, verbs, adverbs, etc. Once you know if a word - perhaps an unfamiliar one - is a noun, you can probably guess you can count it and make it plural if there is more than one (e.g. 1 Tuatara, or 2 Tuataras). If something is a adjective, you can usually change it into an adverb by adding “-ly” (e.g. jejune vs. jejunely). Depending on the context, you may need to decide if a word is in one category or another (e.g “cut” may be a noun when it’s on your finger, or a verb when you are preparing vegetables). These concepts have important analogies when working with R objects.
Exercise: Create objects and check their modes
Create the following objects in R, then use the
mode()function to verify their modes. Try to guess what the mode will be before you look at the solution
chromosome_name <- 'chr02'od_600_value <- 0.47chr_position <- '1001701'spock <- TRUEpilot <- EarhartSolution
Error in eval(expr, envir, enclos): object 'Earhart' not foundmode(chromosome_name)[1] "character"mode(od_600_value)[1] "numeric"mode(chr_position)[1] "character"mode(spock)[1] "logical"mode(pilot)Error in mode(pilot): object 'pilot' not found
Notice from the solution that even if a series of numbers is given as a value
R will consider them to be in the “character” mode if they are enclosed as
single or double quotes. Also, notice that you cannot take a string of alphanumeric
characters (e.g. Earhart) and assign as a value for an object. In this case,
R looks for an object named Earhart but since there is no object, no assignment can
be made. If Earhart did exist, then the mode of pilot would be whatever
the mode of Earhart was originally. If we want to create an object
called pilot that was the name “Earhart”, we need to enclose
Earhart in quotation marks.
pilot <- "Earhart"
mode(pilot)
[1] "character"
Mathematical and functional operations on objects
Once an object exists (which by definition also means it has a mode), R can appropriately manipulate that object. For example, objects of the numeric modes can be added, multiplied, divided, etc. R provides several mathematical (arithmetic) operators including:
| Operator | Description |
|---|---|
| + | addition |
| - | subtraction |
| * | multiplication |
| / | division |
| ^ or ** | exponentiation |
| a%/%b | integer division (division where the remainder is discarded) |
| a%%b | modulus (returns the remainder after division) |
These can be used with literal numbers:
(1 + (5 ** 0.5))/2
[1] 1.618034
and importantly, can be used on any object that evaluates to (i.e. interpreted by R) a numeric object:
# multiply the object 'human_chr_number' by 2
human_chr_number * 2
[1] 46
Exercise: Compute the golden ratio
One approximation of the golden ratio (φ) can be found by taking the sum of 1 and the square root of 5, and dividing by 2 as in the example above. Compute the golden ratio to 3 digits of precision using the
sqrt()andround()functions. Hint: remember theround()function can take 2 arguments.Solution
round((1 + sqrt(5))/2, digits = 3)[1] 1.618Notice that you can place one function inside of another.
Vectors
Vectors are probably the
most used commonly used object type in R.
A vector is a collection of values that are all of the same type (numbers, characters, etc.).
One of the most common
ways to create a vector is to use the c() function - the “concatenate” or
“combine” function. Inside the function you may enter one or more values; for
multiple values, separate each value with a comma:
# Create the SNP gene name vector
snp_genes <- c("OXTR", "ACTN3", "AR", "OPRM1")
Vectors always have a mode and a length.
You can check these with the mode() and length() functions respectively.
Another useful function that gives both of these pieces of information is the
str() (structure) function.
# Check the mode, length, and structure of 'snp_genes'
mode(snp_genes)
[1] "character"
length(snp_genes)
[1] 4
str(snp_genes)
chr [1:4] "OXTR" "ACTN3" "AR" "OPRM1"
Vectors are quite important in R. Another data type that we will work with later in this lesson, data frames, are collections of vectors. What we learn here about vectors will pay off even more when we start working with data frames.
Creating and subsetting vectors
Let’s create a few more vectors to play around with:
# Some interesting human SNPs
# while accuracy is important, typos in the data won't hurt you here
snps <- c('rs53576', 'rs1815739', 'rs6152', 'rs1799971')
snp_chromosomes <- c('3', '11', 'X', '6')
snp_positions <- c(8762685, 66560624, 67545785, 154039662)
Once we have vectors, one thing we may want to do is specifically retrieve one or more values from our vector. To do so, we use bracket notation. We type the name of the vector followed by square brackets. In those square brackets we place the index (e.g. a number) in that bracket as follows:
# get the 3rd value in the snp_genes vector
snp_genes[3]
[1] "AR"
In R, every item your vector is indexed, starting from the first item (1) through to the final number of items in your vector. You can also retrieve a range of numbers:
# get the 1st through 3rd value in the snp_genes vector
snp_genes[1:3]
[1] "OXTR" "ACTN3" "AR"
If you want to retrieve several (but not necessarily sequential) items from a vector, you pass a vector of indices; a vector that has the numbered positions you wish to retrieve.
# get the 1st, 3rd, and 4th value in the snp_genes vector
snp_genes[c(1, 3, 4)]
[1] "OXTR" "AR" "OPRM1"
There are additional (and perhaps less commonly used) ways of subsetting a vector (see these examples). Also, several of these subsetting expressions can be combined:
# get the 1st through the 3rd value, and 4th value in the snp_genes vector
# yes, this is a little silly in a vector of only 4 values.
snp_genes[c(1:3,4)]
[1] "OXTR" "ACTN3" "AR" "OPRM1"
Adding to, removing, or replacing values in existing vectors
Once you have an existing vector, you may want to add a new item to it. To do
so, you can use the c() function again to add your new value:
# add the gene 'CYP1A1' and 'APOA5' to our list of snp genes
# this overwrites our existing vector
snp_genes <- c(snp_genes, "CYP1A1", "APOA5")
We can verify that “snp_genes” contains the new gene entry
snp_genes
[1] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1" "APOA5"
Using a negative index will return a version of a vector with that index’s value removed:
snp_genes[-6]
[1] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1"
We can remove that value from our vector by overwriting it with this expression:
snp_genes <- snp_genes[-6]
snp_genes
[1] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1"
We can also explicitly rename or add a value to our index using double bracket notation:
snp_genes[7]<- "APOA5"
snp_genes
[1] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1" NA "APOA5"
Notice in the operation above that R inserts an NA value to extend our vector so that the gene “APOA5” is an index 7. This may be a good or not-so-good thing depending on how you use this.
Exercise: Examining and subsetting vectors
Answer the following questions to test your knowledge of vectors
Which of the following are true of vectors in R? A) All vectors have a mode or a length
B) All vectors have a mode and a length
C) Vectors may have different lengths
D) Items within a vector may be of different modes
E) You can use thec()to add one or more items to an existing vector
F) You can use thec()to add a vector to an exiting vectorSolution
A) False - Vectors have both of these properties
B) True
C) True
D) False - Vectors have only one mode (e.g. numeric, character); all items in
a vector must be of this mode. E) True
F) True
Logical Subsetting
There is one last set of cool subsetting capabilities we want to introduce. It is possible within R to retrieve items in a vector based on a logical evaluation or numerical comparison. For example, let’s say we wanted get all of the SNPs in our vector of SNP positions that were greater than 100,000,000. We could index using the ‘>’ (greater than) logical operator:
snp_positions[snp_positions > 100000000]
[1] 154039662
In the square brackets you place the name of the vector followed by the comparison operator and (in this case) a numeric value. Some of the most common logical operators you will use in R are:
| Operator | Description |
|---|---|
| < | less than |
| <= | less than or equal to |
| > | greater than |
| >= | greater than or equal to |
| == | exactly equal to |
| != | not equal to |
| !x | not x |
| a | b | a or b |
| a & b | a and b |
The magic of programming
The reason why the expression
snp_positions[snp_positions > 100000000]works can be better understood if you examine what the expression “snp_positions > 100000000” evaluates to:snp_positions > 100000000[1] FALSE FALSE FALSE TRUEThe output above is a logical vector, the 4th element of which is TRUE. When you pass a logical vector as an index, R will return the true values:
snp_positions[c(FALSE, FALSE, FALSE, TRUE)][1] 154039662If you have never coded before, this type of situation starts to expose the “magic” of programming. We mentioned before that in the bracket notation you take your named vector followed by brackets which contain an index: named_vector[index]. The “magic” is that the index needs to evaluate to a number. So, even if it does not appear to be an integer (e.g. 1, 2, 3), as long as R can evaluate it, we will get a result. That our expression
snp_positions[snp_positions > 100000000]evaluates to a number can be seen in the following situation. If you wanted to know which index (1, 2, 3, or 4) in our vector of SNP positions was the one that was greater than 100,000,000?We can use the
which()function to return the indices of any item that evaluates as TRUE in our comparison:which(snp_positions > 100000000)[1] 4Why this is important
Often in programming we will not know what inputs and values will be used when our code is executed. Rather than put in a pre-determined value (e.g 100000000) we can use an object that can take on whatever value we need. So for example:
snp_marker_cutoff <- 100000000 snp_positions[snp_positions > snp_marker_cutoff][1] 154039662Ultimately, it’s putting together flexible, reusable code like this that gets at the “magic” of programming!
A few final vector tricks
Finally, there are a few other common retrieve or replace operations you may
want to know about. First, you can check to see if any of the values of your
vector are missing (i.e. are NA). Missing data will get a more detailed treatment later,
but the is.NA() function will return a logical vector, with TRUE for any NA
value:
# current value of 'snp_genes':
# chr [1:7] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1" NA "APOA5"
is.na(snp_genes)
[1] FALSE FALSE FALSE FALSE FALSE TRUE FALSE
Sometimes, you may wish to find out if a specific value (or several values) is
present a vector. You can do this using the comparison operator %in%, which
will return TRUE for any value in your collection that is in
the vector you are searching:
# current value of 'snp_genes':
# chr [1:7] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1" NA "APOA5"
# test to see if "ACTN3" or "APO5A" is in the snp_genes vector
# if you are looking for more than one value, you must pass this as a vector
c("ACTN3","APOA5") %in% snp_genes
[1] TRUE TRUE
Review Exercise 1
What data types/modes are the following vectors? a.
snps
b.snp_chromosomes
c.snp_positionsSolution
typeof(snps)[1] "character"typeof(snp_chromosomes)[1] "character"typeof(snp_positions)[1] "double"
Review Exercise 2
Add the following values to the specified vectors: a. To the
snpsvector add: ‘rs662799’
b. To thesnp_chromosomesvector add: 11
c. To thesnp_positionsvector add: 116792991Solution
snps <- c(snps, 'rs662799') snps[1] "rs53576" "rs1815739" "rs6152" "rs1799971" "rs662799"snp_chromosomes <- c(snp_chromosomes, "11") # did you use quotes? snp_chromosomes[1] "3" "11" "X" "6" "11"snp_positions <- c(snp_positions, 116792991) snp_positions[1] 8762685 66560624 67545785 154039662 116792991
Review Exercise 3
Make the following change to the
snp_genesvector:Hint: Your vector should look like this in ‘Environment’:
chr [1:7] "OXTR" "ACTN3" "AR" "OPRM1" "CYP1A1" NA "APOA5". If not recreate the vector by running this expression:snp_genes <- c("OXTR", "ACTN3", "AR", "OPRM1", "CYP1A1", NA, "APOA5")a. Create a new version of
snp_genesthat does not contain CYP1A1 and then
b. Add 2 NA values to the end ofsnp_genesSolution
snp_genes <- snp_genes[-5] snp_genes <- c(snp_genes, NA, NA) snp_genes[1] "OXTR" "ACTN3" "AR" "OPRM1" NA "APOA5" NA NA
Review Exercise 4
Using indexing, create a new vector named
combinedthat contains:
- The the 1st value in
snp_genes- The 1st value in
snps- The 1st value in
snp_chromosomes- The 1st value in
snp_positionsSolution
combined <- c(snp_genes[1], snps[1], snp_chromosomes[1], snp_positions[1]) combined[1] "OXTR" "rs53576" "3" "8762685"
Review Exercise 5
What type of data is
combined?Solution
typeof(combined)[1] "character"
Bonus material: Lists
Lists are quite useful in R, but we won’t be using them in the genomics lessons. That said, you may come across lists in the way that some bioinformatics programs may store and/or return data to you. One of the key attributes of a list is that, unlike a vector, a list may contain data of more than one mode. Learn more about creating and using lists using this nice tutorial. In this one example, we will create a named list and show you how to retrieve items from the list.
# Create a named list using the 'list' function and our SNP examples
# Note, for easy reading we have placed each item in the list on a separate line
# Nothing special about this, you can do this for any multiline commands
# To run this command, make sure the entire command (all 4 lines) are highlighted
# before running
# Note also, as we are doing all this inside the list() function use of the
# '=' sign is good style
snp_data <- list(genes = snp_genes,
refference_snp = snps,
chromosome = snp_chromosomes,
position = snp_positions)
# Examine the structure of the list
str(snp_data)
List of 4
$ genes : chr [1:8] "OXTR" "ACTN3" "AR" "OPRM1" ...
$ refference_snp: chr [1:5] "rs53576" "rs1815739" "rs6152" "rs1799971" ...
$ chromosome : chr [1:5] "3" "11" "X" "6" ...
$ position : num [1:5] 8.76e+06 6.66e+07 6.75e+07 1.54e+08 1.17e+08
To get all the values for the position object in the list, we use the $ notation:
# return all the values of position object
snp_data$position
[1] 8762685 66560624 67545785 154039662 116792991
To get the first value in the position object, use the [] notation to index:
# return first value of the position object
snp_data$position[1]
[1] 8762685
Key Points
Effectively using R is a journey of months or years. Still you don’t have to be an expert to use R and you can start using and analyzing your data with with about a day’s worth of training
It is important to understand how data are organized by R in a given object type and how the mode of that type (e.g. numeric, character, logical, etc.) will determine how R will operate on that data.
Working with vectors effectively prepares you for understanding how data are organized in R.
Introduction to the example dataset and file type
Overview
Teaching: 15 min
Exercises: 0 minQuestions
What data are we using in the lesson?
What are VCF files?
Objectives
Know what the example dataset represents
Know the concepts of how VCF files are generated
Preface
The Intro to R and RStudio for Genomics is a part of the Genomics Data Carpentry lessons. In this lesson we will learn the necessary skill sets for R and RStudio and apply them directly to a real next-generation sequencing (NGS) data in the variant calling format (VCF) file type. Previous Genomics Data Carpentry lessons teach learners how to generate a VCF file from FASTQ files downloaded from NCBI Sequence Read Archive (SRA), so we won’t cover that here. Instead, in this episode we will give a brief overview of the data and a what VCF file types are for those who wish to teach the Intro to R and RStudio for Genomics lesson independently of the Genomics Data Carpentry lessons.
This dataset was selected for several reasons, including:
- Simple, but iconic NGS-problem: Examine a population where we want to characterize changes in sequence a priori
- Dataset publicly available - in this case through the NCBI SRA (http://www.ncbi.nlm.nih.gov/sra)
Introduction to the dataset
Microbes are ideal organisms for exploring ‘Long-term Evolution Experiments’ (LTEEs) - thousands of generations can be generated and stored in a way that would be virtually impossible for more complex eukaryotic systems. In Tenaillon et al 2016, 12 populations of Escherichia coli were propagated for more than 50,000 generations in a glucose-limited minimal medium. This medium was supplemented with citrate which E. coli cannot metabolize in the aerobic conditions of the experiment. Sequencing of the populations at regular time points reveals that spontaneous citrate-using mutants (Cit+) appeared in a population of E.coli (designated Ara-3) at around 31,000 generations. It should be noted that spontaneous Cit+ mutants are extraordinarily rare - inability to metabolize citrate is one of the defining characters of the E. coli species. Eventually, Cit+ mutants became the dominant population as the experimental growth medium contained a high concentration of citrate relative to glucose. Around the same time that this mutation emerged, another phenotype become prominent in the Ara-3 population. Many E. coli began to develop excessive numbers of mutations, meaning they became hypermutable.
Strains from generation 0 to generation 50,000 were sequenced, including ones that were both Cit+ and Cit- and hypermutable in later generations.
For the purposes of this workshop we’re going to be working with 3 of the sequence reads from this experiment.
| SRA Run Number | Clone | Generation | Cit | Hypermutable | Read Length | Sequencing Depth |
|---|---|---|---|---|---|---|
| SRR2589044 | REL2181A | 5,000 | Unknown | None | 150 | 60.2 |
| SRR2584863 | REL7179B | 15,000 | Unknown | None | 150 | 88 |
| SRR2584866 | REL11365 | 50,000 | Cit+ | plus | 150 | 138.3 |
We want to be able to look at differences in mutation rates between hypermutable and non-hypermutable strains. We also want to analyze the sequences to figure out what changes occurred in genomes to make the strains Cit+. Ultimately, we will use R to answer the questions:
- How many base pair changes are there between the Cit+ and Cit- strains?
- What are the base pair changes between strains?
How VCF files are generated
Publicly accessible sequencing files in FASTQ formats can be downloaded from NCBI SRA. However, at FASTQ files contain unaligned sequences of varying quality, and requires clean up and alignment steps for variants to be called from the reference genome.
Five steps are taken to transform FASTQ files to variant calls contained in VCF files and at each step, specialized non-R based bioinformatics tools that are used:
How variant calls are stored in VCF files
VCF files contain variants that were called against a reference genome. These files are slightly more complicated than regular tables you can open using programs like Excel and contain two sections: header and records.
Below you will see the header (which describes the format), the time and date the file was created, the version of bcftools that was used, the command line parameters used, and some additional information:
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##bcftoolsVersion=1.8+htslib-1.8
##bcftoolsCommand=mpileup -O b -o results/bcf/SRR2584866_raw.bcf -f data/ref_genome/ecoli_rel606.fasta results/bam/SRR2584866.aligned.sorted.bam
##reference=file://data/ref_genome/ecoli_rel606.fasta
##contig=<ID=CP000819.1,length=4629812>
##ALT=<ID=*,Description="Represents allele(s) other than observed.">
##INFO=<ID=INDEL,Number=0,Type=Flag,Description="Indicates that the variant is an INDEL.">
##INFO=<ID=IDV,Number=1,Type=Integer,Description="Maximum number of reads supporting an indel">
##INFO=<ID=IMF,Number=1,Type=Float,Description="Maximum fraction of reads supporting an indel">
##INFO=<ID=DP,Number=1,Type=Integer,Description="Raw read depth">
##INFO=<ID=VDB,Number=1,Type=Float,Description="Variant Distance Bias for filtering splice-site artefacts in RNA-seq data (bigger is better)",Version=
##INFO=<ID=RPB,Number=1,Type=Float,Description="Mann-Whitney U test of Read Position Bias (bigger is better)">
##INFO=<ID=MQB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality Bias (bigger is better)">
##INFO=<ID=BQB,Number=1,Type=Float,Description="Mann-Whitney U test of Base Quality Bias (bigger is better)">
##INFO=<ID=MQSB,Number=1,Type=Float,Description="Mann-Whitney U test of Mapping Quality vs Strand Bias (bigger is better)">
##INFO=<ID=SGB,Number=1,Type=Float,Description="Segregation based metric.">
##INFO=<ID=MQ0F,Number=1,Type=Float,Description="Fraction of MQ0 reads (smaller is better)">
##FORMAT=<ID=PL,Number=G,Type=Integer,Description="List of Phred-scaled genotype likelihoods">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##INFO=<ID=ICB,Number=1,Type=Float,Description="Inbreeding Coefficient Binomial test (bigger is better)">
##INFO=<ID=HOB,Number=1,Type=Float,Description="Bias in the number of HOMs number (smaller is better)">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes for each ALT allele, in the same order as listed">
##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes">
##INFO=<ID=DP4,Number=4,Type=Integer,Description="Number of high-quality ref-forward , ref-reverse, alt-forward and alt-reverse bases">
##INFO=<ID=MQ,Number=1,Type=Integer,Description="Average mapping quality">
##bcftools_callVersion=1.8+htslib-1.8
##bcftools_callCommand=call --ploidy 1 -m -v -o results/bcf/SRR2584866_variants.vcf results/bcf/SRR2584866_raw.bcf; Date=Tue Oct 9 18:48:10 2018
Followed by information on each of the variations observed:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT results/bam/SRR2584866.aligned.sorted.bam
CP000819.1 1521 . C T 207 . DP=9;VDB=0.993024;SGB=-0.662043;MQSB=0.974597;MQ0F=0;AC=1;AN=1;DP4=0,0,4,5;MQ=60
CP000819.1 1612 . A G 225 . DP=13;VDB=0.52194;SGB=-0.676189;MQSB=0.950952;MQ0F=0;AC=1;AN=1;DP4=0,0,6,5;MQ=60
CP000819.1 9092 . A G 225 . DP=14;VDB=0.717543;SGB=-0.670168;MQSB=0.916482;MQ0F=0;AC=1;AN=1;DP4=0,0,7,3;MQ=60
CP000819.1 9972 . T G 214 . DP=10;VDB=0.022095;SGB=-0.670168;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,2,8;MQ=60 GT:PL
CP000819.1 10563 . G A 225 . DP=11;VDB=0.958658;SGB=-0.670168;MQSB=0.952347;MQ0F=0;AC=1;AN=1;DP4=0,0,5,5;MQ=60
CP000819.1 22257 . C T 127 . DP=5;VDB=0.0765947;SGB=-0.590765;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,2,3;MQ=60 GT:PL
CP000819.1 38971 . A G 225 . DP=14;VDB=0.872139;SGB=-0.680642;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,4,8;MQ=60 GT:PL
CP000819.1 42306 . A G 225 . DP=15;VDB=0.969686;SGB=-0.686358;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,5,9;MQ=60 GT:PL
CP000819.1 45277 . A G 225 . DP=15;VDB=0.470998;SGB=-0.680642;MQSB=0.95494;MQ0F=0;AC=1;AN=1;DP4=0,0,7,5;MQ=60
CP000819.1 56613 . C G 183 . DP=12;VDB=0.879703;SGB=-0.676189;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,8,3;MQ=60 GT:PL
CP000819.1 62118 . A G 225 . DP=19;VDB=0.414981;SGB=-0.691153;MQSB=0.906029;MQ0F=0;AC=1;AN=1;DP4=0,0,8,10;MQ=59
CP000819.1 64042 . G A 225 . DP=18;VDB=0.451328;SGB=-0.689466;MQSB=1;MQ0F=0;AC=1;AN=1;DP4=0,0,7,9;MQ=60 GT:PL
The first few columns represent the information we have about a predicted variation.
| column | info |
|---|---|
| CHROM | contig location where the variation occurs |
| POS | position within the contig where the variation occurs |
| ID | a . until we add annotation information |
| REF | reference genotype (forward strand) |
| ALT | sample genotype (forward strand) |
| QUAL | Phred-scaled probability that the observed variant exists at this site (higher is better) |
| FILTER | a . if no quality filters have been applied, PASS if a filter is passed, or the name of the filters this variant failed |
In an ideal world, the information in the QUAL column would be all we needed to filter out bad variant calls.
However, in reality we need to filter on multiple other metrics.
The last two columns contain the genotypes and can be tricky to decode.
| column | info |
|---|---|
| FORMAT | lists in order the metrics presented in the final column |
| results | lists the values associated with those metrics in order |
For our file, the metrics presented are GT:PL:GQ.
| metric | definition |
|---|---|
| AD, DP | the depth per allele by sample and coverage |
| GT | the genotype for the sample at this loci. For a diploid organism, the GT field indicates the two alleles carried by the sample, encoded by a 0 for the REF allele, 1 for the first ALT allele, 2 for the second ALT allele, etc. A 0/0 means homozygous reference, 0/1 is heterozygous, and 1/1 is homozygous for the alternate allele. |
| PL | the likelihoods of the given genotypes |
| GQ | the Phred-scaled confidence for the genotype |
For more information on VCF files visit The Broad Institute’s VCF guide.
References
Tenaillon O, Barrick JE, Ribeck N, Deatherage DE, Blanchard JL, Dasgupta A, Wu GC, Wielgoss S, Cruveiller S, Médigue C, Schneider D, Lenski RE. Tempo and mode of genome evolution in a 50,000-generation experiment (2016) Nature. 536(7615): 165–170. Paper, Supplemental materials Data on NCBI SRA: https://trace.ncbi.nlm.nih.gov/Traces/sra/?study=SRP064605 Data on EMBL-EBI ENA: https://www.ebi.ac.uk/ena/data/view/PRJNA295606
This episode was adapted from the Data Carpentry Genomic lessons:
Key Points
The dataset comes from a real world experiment in E. coli.
Publicly available FASTQ files can be downloaded from NCBI SRA.
Several steps are taken outside of R/RStudio to create VCF files from FASTQ files.
VCF files store variant calls in a special format.
R Basics continued - factors and data frames
Overview
Teaching: 60 min
Exercises: 30 minQuestions
How do I get started with tabular data (e.g. spreadsheets) in R?
What are some best practices for reading data into R?
How do I save tabular data generated in R?
Objectives
Explain the basic principle of tidy datasets
Be able to load a tabular dataset using base R functions
Be able to determine the structure of a data frame including its dimensions and the datatypes of variables
Be able to subset/retrieve values from a data frame
Understand how R may coerce data into different modes
Be able to change the mode of an object
Understand that R uses factors to store and manipulate categorical data
Be able to manipulate a factor, including subsetting and reordering
Be able to apply an arithmetic function to a data frame
Be able to coerce the class of an object (including variables in a data frame)
Be able to import data from Excel
Be able to save a data frame as a delimited file
Working with spreadsheets (tabular data)
A substantial amount of the data we work with in genomics will be tabular data, this is data arranged in rows and columns - also known as spreadsheets. We could write a whole lesson on how to work with spreadsheets effectively (actually we did). For our purposes, we want to remind you of a few principles before we work with our first set of example data:
1) Keep raw data separate from analyzed data
This is principle number one because if you can’t tell which files are the original raw data, you risk making some serious mistakes (e.g. drawing conclusion from data which have been manipulated in some unknown way).
2) Keep spreadsheet data Tidy
The simplest principle of Tidy data is that we have one row in our spreadsheet for each observation or sample, and one column for every variable that we measure or report on. As simple as this sounds, it’s very easily violated. Most data scientists agree that significant amounts of their time is spent tidying data for analysis. Read more about data organization in our lesson and in this paper.
3) Trust but verify
Finally, while you don’t need to be paranoid about data, you should have a plan for how you will prepare it for analysis. This a focus of this lesson. You probably already have a lot of intuition, expectations, assumptions about your data - the range of values you expect, how many values should have been recorded, etc. Of course, as the data get larger our human ability to keep track will start to fail (and yes, it can fail for small data sets too). R will help you to examine your data so that you can have greater confidence in your analysis, and its reproducibility.
Tip: Keeping you raw data separate
When you work with data in R, you are not changing the original file you loaded that data from. This is different than (for example) working with a spreadsheet program where changing the value of the cell leaves you one “save”-click away from overwriting the original file. You have to purposely use a writing function (e.g.
write.csv()) to save data loaded into R. In that case, be sure to save the manipulated data into a new file. More on this later in the lesson.
Importing tabular data into R
There are several ways to import data into R. For our purpose here, we will
focus on using the tools every R installation comes with (so called “base” R) to
import a comma-delimited file containing the results of our variant calling workflow.
We will need to load the sheet using a function called read.csv().
Exercise: Review the arguments of the
read.csv()functionBefore using the
read.csv()function, use R’s help feature to answer the following questions.Hint: Entering ‘?’ before the function name and then running that line will bring up the help documentation. Also, when reading this particular help be careful to pay attention to the ‘read.csv’ expression under the ‘Usage’ heading. Other answers will be in the ‘Arguments’ heading.
A) What is the default parameter for ‘header’ in the
read.csv()function?B) What argument would you have to change to read a file that was delimited by semicolons (;) rather than commas?
C) What argument would you have to change to read file in which numbers used commas for decimal separation (i.e. 1,00)?
D) What argument would you have to change to read in only the first 10,000 rows of a very large file?
Solution
A) The
read.csv()function has the argument ‘header’ set to TRUE by default, this means the function always assumes the first row is header information, (i.e. column names)B) The
read.csv()function has the argument ‘sep’ set to “,”. This means the function assumes commas are used as delimiters, as you would expect. Changing this parameter (e.g.sep=";") would now interpret semicolons as delimiters.C) Although it is not listed in the
read.csv()usage,read.csv()is a “version” of the functionread.table()and accepts all its arguments. If you setdec=","you could change the decimal operator. We’d probably assume the delimiter is some other character.D) You can set
nrowto a numeric value (e.g.nrow=10000) to choose how many rows of a file you read in. This may be useful for very large files where not all the data is needed to test some data cleaning steps you are applying.Hopefully, this exercise gets you thinking about using the provided help documentation in R. There are many arguments that exist, but which we wont have time to cover. Look here to get familiar with functions you use frequently, you may be surprised at what you find they can do.
Now, let’s read in the file combined_tidy_vcf.csv which will be located in
/home/dcuser/r_data/. Call this data variants. The
first argument to pass to our read.csv() function is the file path for our
data. The file path must be in quotes and now is a good time to remember to
use tab autocompletion. If you use tab autocompletion you avoid typos and
errors in file paths. Use it!
## read in a CSV file and save it as 'variants'
variants <- read.csv("/home/dcuser/r_data/combined_tidy_vcf.csv")
One of the first things you should notice is that in the Environment window,
you have the variants object, listed as 801 obs. (observations/rows)
of 29 variables (columns). Double-clicking on the name of the object will open
a view of the data in a new tab.
Summarizing, subsetting, and determining the structure of a data frame.
A data frame is the standard way in R to store tabular data. A data fame could also be thought of as a collection of vectors, all of which have the same length. Using only two functions, we can learn a lot about out data frame including some summary statistics as well as well as the “structure” of the data frame. Let’s examine what each of these functions can tell us:
## get summary statistics on a data frame
summary(variants)
sample_id CHROM POS ID
Length:801 Length:801 Min. : 1521 Mode:logical
Class :character Class :character 1st Qu.:1115970 NA's:801
Mode :character Mode :character Median :2290361
Mean :2243682
3rd Qu.:3317082
Max. :4629225
REF ALT QUAL FILTER
Length:801 Length:801 Min. : 4.385 Mode:logical
Class :character Class :character 1st Qu.:139.000 NA's:801
Mode :character Mode :character Median :195.000
Mean :172.276
3rd Qu.:225.000
Max. :228.000
INDEL IDV IMF DP
Mode :logical Min. : 2.000 Min. :0.5714 Min. : 2.00
FALSE:700 1st Qu.: 7.000 1st Qu.:0.8824 1st Qu.: 7.00
TRUE :101 Median : 9.000 Median :1.0000 Median :10.00
Mean : 9.396 Mean :0.9219 Mean :10.57
3rd Qu.:11.000 3rd Qu.:1.0000 3rd Qu.:13.00
Max. :20.000 Max. :1.0000 Max. :79.00
NA's :700 NA's :700
VDB RPB MQB BQB
Min. :0.0005387 Min. :0.0000 Min. :0.0000 Min. :0.1153
1st Qu.:0.2180410 1st Qu.:0.3776 1st Qu.:0.1070 1st Qu.:0.6963
Median :0.4827410 Median :0.8663 Median :0.2872 Median :0.8615
Mean :0.4926291 Mean :0.6970 Mean :0.5330 Mean :0.7784
3rd Qu.:0.7598940 3rd Qu.:1.0000 3rd Qu.:1.0000 3rd Qu.:1.0000
Max. :0.9997130 Max. :1.0000 Max. :1.0000 Max. :1.0000
NA's :773 NA's :773 NA's :773
MQSB SGB MQ0F ICB
Min. :0.01348 Min. :-0.6931 Min. :0.00000 Mode:logical
1st Qu.:0.95494 1st Qu.:-0.6762 1st Qu.:0.00000 NA's:801
Median :1.00000 Median :-0.6620 Median :0.00000
Mean :0.96428 Mean :-0.6444 Mean :0.01127
3rd Qu.:1.00000 3rd Qu.:-0.6364 3rd Qu.:0.00000
Max. :1.01283 Max. :-0.4536 Max. :0.66667
NA's :48
HOB AC AN DP4 MQ
Mode:logical Min. :1 Min. :1 Length:801 Min. :10.00
NA's:801 1st Qu.:1 1st Qu.:1 Class :character 1st Qu.:60.00
Median :1 Median :1 Mode :character Median :60.00
Mean :1 Mean :1 Mean :58.19
3rd Qu.:1 3rd Qu.:1 3rd Qu.:60.00
Max. :1 Max. :1 Max. :60.00
Indiv gt_PL gt_GT gt_GT_alleles
Length:801 Length:801 Min. :1 Length:801
Class :character Class :character 1st Qu.:1 Class :character
Mode :character Mode :character Median :1 Mode :character
Mean :1
3rd Qu.:1
Max. :1
Our data frame had 29 variables, so we get 29 fields that summarize the data.
The QUAL, IMF, and VDB variables (and several others) are
numerical data and so you get summary statistics on the min and max values for
these columns, as well as mean, median, and interquartile ranges. Many of the
other variables (e.g. sample_id) are treated as characters data (more on this
in a bit).
There is a lot to work with, so we will subset the first three columns into a
new data frame using the data.frame() function.
## put the first three columns of variants into a new data frame called subset
subset<-data.frame(variants[,c(1:3,6)])
Now, let’s use the str() (structure) function to look a little more closely
at how data frames work:
## get the structure of a data frame
str(subset)
'data.frame': 801 obs. of 4 variables:
$ sample_id: chr "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863" ...
$ CHROM : chr "CP000819.1" "CP000819.1" "CP000819.1" "CP000819.1" ...
$ POS : int 9972 263235 281923 433359 473901 648692 1331794 1733343 2103887 2333538 ...
$ ALT : chr "G" "T" "T" "CTTTTTTTT" ...
Ok, thats a lot up unpack! Some things to notice.
- the object type
data.frameis displayed in the first row along with its dimensions, in this case 801 observations (rows) and 4 variables (columns) - Each variable (column) has a name (e.g.
sample_id). This is followed by the object mode (e.g. chr, int, etc.). Notice that before each variable name there is a$- this will be important later.
Introducing Factors
Factors are the final major data structure we will introduce in our R genomics lessons. Factors can be thought of as vectors which are specialized for categorical data. Given R’s specialization for statistics, this make sense since categorial and continuous variables are usually treated differently. Sometimes you may want to have data treated as a factor, but in other cases, this may be undesirable.
Let’s see the value of treating some of which are categorical in nature as factors. Let’s take a look at just the alternate alleles
## extract the "ALT" column to a new object
alt_alleles <- subset$ALT
Let’s look at the first few items in our factor using head():
head(alt_alleles)
[1] "G" "T" "T" "CTTTTTTTT" "CCGCGC" "T"
There are 801 alleles (one for each row). To simplify, lets look at just the single-nuleotide alleles (SNPs). We can use some of the vector indexing skills from the last episode.
snps <- c(alt_alleles[alt_alleles=="A"],
alt_alleles[alt_alleles=="T"],
alt_alleles[alt_alleles=="G"],
alt_alleles[alt_alleles=="C"])
This leaves us with a vector of the 701 alternative alleles which were single nucleotides. Right now, they are being treated a characters, but we could treat them as categories of SNP. Doing this will enable some nice features. For example, we can try to generate a plot of this character vector as it is right now:
plot(snps)
Warning in xy.coords(x, y, xlabel, ylabel, log): NAs introduced by coercion
Warning in min(x): no non-missing arguments to min; returning Inf
Warning in max(x): no non-missing arguments to max; returning -Inf
Error in plot.window(...): need finite 'ylim' values
Whoops! Though the plot() function will do its best to give us a quick plot,
it is unable to do so here. One way to fix this it to tell R to treat the SNPs
as categories (i.e. a factor vector); we will create a new object to avoid
confusion using the factor() function:
factor_snps <- factor(snps)
Let’s learn a little more about this new type of vector:
str(factor_snps)
Factor w/ 4 levels "A","C","G","T": 1 1 1 1 1 1 1 1 1 1 ...
What we get back are the categories (“A”,”C”,”G”,”T”) in our factor; these are called “Levels”. Levels are the different categories contained in a factor. By default, R will organize the levels in a factor in alphabetical order. So the first level in this factor is “A”.
For the sake of efficiency, R stores the content of a factor as a vector of
integers, which an integer is assigned to each of the possible levels. Recall
levels are assigned in alphabetical order. In this case, the first item in our
factor_snps object is “A”, which happens to be the 1st level of our factor,
ordered alphabetically. This explains the sequence of “1”s (“Factor w/ 4 levels
“A”,”C”,”G”,”T”: 1 1 1 1 1 1 1 1 1 1 …”), since “A” is the first level, and
the first few items in our factor are all “A”s.
We can see how many items in our vector fall into each category:
summary(factor_snps)
A C G T
211 139 154 203
As you can imagine, this is already useful when you want to generate a tally.
Tip: treating objects as categories without changing their mode
You don’t have to make an object a factor to get the benefits of treating an object as a factor. See what happens when you use the
as.factor()function onfactor_snps. To generate a tally, you can sometimes also use thetable()function; though sometimes you may need to combine both (i.e.table(as.factor(object)))
Plotting and ordering factors
One of the most common uses for factors will be when you plot categorical values. For example, suppose we want to know how many of our variants had each possible SNP we could generate a plot:
plot(factor_snps)
This isn’t a particularly pretty example of a plot but it works. We’ll be learning much more about creating nice, publication-quality graphics later in this lesson.
If you recall, factors are ordered alphabetically. That might make sense, but categories (e.g., “red”, “blue”, “green”) often do not have an intrinsic order. What if we wanted to order our plot according to the numerical value (i.e., in descending order of SNP frequency)? We can enforce an order on our factors:
ordered_factor_snps <- factor(factor_snps, levels = names(sort(table(factor_snps))))
Let’s deconstruct this from the inside out (you can try each of these commands to see why this works):
- We create a table of
factor_snpsto get the frequency of each SNP:table(factor_snps) - We sort this table:
sort(table(factor_snps)); use thedecreasing =parameter for this function if you wanted to change from the default of FALSE - Using the
namesfunction gives us just the character names of the table sorted by frequencies:names(sort(table(factor_snps))) - The
factorfunction is what allows us to create a factor. We give it thefactor_snpsobject as input, and use thelevels=parameter to enforce the ordering of the levels.
Now we see our plot has be reordered:
plot(ordered_factor_snps)
Factors come in handy in many places when using R. Even using more sophisticated plotting packages such as ggplot2 will sometimes require you to understand how to manipulate factors.
Subsetting data frames
Next, we are going to talk about how you can get specific values from data frames, and where necessary, change the mode of a column of values.
The first thing to remember is that a data frame is two-dimensional (rows and
columns). Therefore, to select a specific value we will will once again use
[] (bracket) notation, but we will specify more than one value (except in some cases
where we are taking a range).
Exercise: Subsetting a data frame
Try the following indices and functions and try to figure out what they return
a.
variants[1,1]b.
variants[2,4]c.
variants[801,29]d.
variants[2, ]e.
variants[-1, ]f.
variants[1:4,1]g.
variants[1:10,c("REF","ALT")]h.
variants[,c("sample_id")]i.
head(variants)j.
tail(variants)k.
variants$sample_idl.
variants[variants$REF == "A",]Solution
a.
variants[1,1][1] "SRR2584863"b.
variants[2,4][1] NAc.
variants[801,29][1] "T"d.
variants[2, ]sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP VDB 2 SRR2584863 CP000819.1 263235 NA G T 85 NA FALSE NA NA 6 0.096133 RPB MQB BQB MQSB SGB MQ0F ICB HOB AC AN DP4 MQ 2 1 1 1 NA -0.590765 0.166667 NA NA 1 1 0,1,0,5 33 Indiv gt_PL 2 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 112,0 gt_GT gt_GT_alleles 2 1 Te.
variants[-1, ]sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF 2 SRR2584863 CP000819.1 263235 NA G T 85 NA FALSE NA NA 3 SRR2584863 CP000819.1 281923 NA G T 217 NA FALSE NA NA 4 SRR2584863 CP000819.1 433359 NA CTTTTTTT CTTTTTTTT 64 NA TRUE 12 1.0 5 SRR2584863 CP000819.1 473901 NA CCGC CCGCGC 228 NA TRUE 9 0.9 6 SRR2584863 CP000819.1 648692 NA C T 210 NA FALSE NA NA 7 SRR2584863 CP000819.1 1331794 NA C A 178 NA FALSE NA NA DP VDB RPB MQB BQB MQSB SGB MQ0F ICB HOB AC AN DP4 MQ 2 6 0.096133 1 1 1 NA -0.590765 0.166667 NA NA 1 1 0,1,0,5 33 3 10 0.774083 NA NA NA 0.974597 -0.662043 0.000000 NA NA 1 1 0,0,4,5 60 4 12 0.477704 NA NA NA 1.000000 -0.676189 0.000000 NA NA 1 1 0,1,3,8 60 5 10 0.659505 NA NA NA 0.916482 -0.662043 0.000000 NA NA 1 1 1,0,2,7 60 6 10 0.268014 NA NA NA 0.916482 -0.670168 0.000000 NA NA 1 1 0,0,7,3 60 7 8 0.624078 NA NA NA 0.900802 -0.651104 0.000000 NA NA 1 1 0,0,3,5 60 Indiv gt_PL 2 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 112,0 3 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 247,0 4 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 91,0 5 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 255,0 6 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 240,0 7 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 208,0 gt_GT gt_GT_alleles 2 1 T 3 1 T 4 1 CTTTTTTTT 5 1 CCGCGC 6 1 T 7 1 Af.
variants[1:4,1][1] "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863"g.
variants[1:10,c("REF","ALT")]REF 1 T 2 G 3 G 4 CTTTTTTT 5 CCGC 6 C 7 C 8 G 9 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 10 AT ALT 1 G 2 T 3 T 4 CTTTTTTTT 5 CCGCGC 6 T 7 A 8 A 9 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 10 ATTh.
variants[,c("sample_id")][1] "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863" [6] "SRR2584863"i.
head(variants)sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF 1 SRR2584863 CP000819.1 9972 NA T G 91 NA FALSE NA NA 2 SRR2584863 CP000819.1 263235 NA G T 85 NA FALSE NA NA 3 SRR2584863 CP000819.1 281923 NA G T 217 NA FALSE NA NA 4 SRR2584863 CP000819.1 433359 NA CTTTTTTT CTTTTTTTT 64 NA TRUE 12 1.0 5 SRR2584863 CP000819.1 473901 NA CCGC CCGCGC 228 NA TRUE 9 0.9 6 SRR2584863 CP000819.1 648692 NA C T 210 NA FALSE NA NA DP VDB RPB MQB BQB MQSB SGB MQ0F ICB HOB AC AN DP4 MQ 1 4 0.0257451 NA NA NA NA -0.556411 0.000000 NA NA 1 1 0,0,0,4 60 2 6 0.0961330 1 1 1 NA -0.590765 0.166667 NA NA 1 1 0,1,0,5 33 3 10 0.7740830 NA NA NA 0.974597 -0.662043 0.000000 NA NA 1 1 0,0,4,5 60 4 12 0.4777040 NA NA NA 1.000000 -0.676189 0.000000 NA NA 1 1 0,1,3,8 60 5 10 0.6595050 NA NA NA 0.916482 -0.662043 0.000000 NA NA 1 1 1,0,2,7 60 6 10 0.2680140 NA NA NA 0.916482 -0.670168 0.000000 NA NA 1 1 0,0,7,3 60 Indiv gt_PL 1 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 121,0 2 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 112,0 3 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 247,0 4 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 91,0 5 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 255,0 6 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 240,0 gt_GT gt_GT_alleles 1 1 G 2 1 T 3 1 T 4 1 CTTTTTTTT 5 1 CCGCGC 6 1 Tj.
tail(variants)sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP 796 SRR2589044 CP000819.1 3444175 NA G T 184 NA FALSE NA NA 9 797 SRR2589044 CP000819.1 3481820 NA A G 225 NA FALSE NA NA 12 798 SRR2589044 CP000819.1 3893550 NA AG AGG 101 NA TRUE 4 1 4 799 SRR2589044 CP000819.1 3901455 NA A AC 70 NA TRUE 3 1 3 800 SRR2589044 CP000819.1 4100183 NA A G 177 NA FALSE NA NA 8 801 SRR2589044 CP000819.1 4431393 NA TGG T 225 NA TRUE 10 1 10 VDB RPB MQB BQB MQSB SGB MQ0F ICB HOB AC AN DP4 MQ 796 0.4714620 NA NA NA 0.992367 -0.651104 0 NA NA 1 1 0,0,4,4 60 797 0.8707240 NA NA NA 1.000000 -0.680642 0 NA NA 1 1 0,0,4,8 60 798 0.9182970 NA NA NA 1.000000 -0.556411 0 NA NA 1 1 0,0,3,1 52 799 0.0221621 NA NA NA NA -0.511536 0 NA NA 1 1 0,0,3,0 60 800 0.9272700 NA NA NA 0.900802 -0.651104 0 NA NA 1 1 0,0,3,5 60 801 0.7488140 NA NA NA 1.007750 -0.670168 0 NA NA 1 1 0,0,4,6 60 Indiv gt_PL 796 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 214,0 797 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 255,0 798 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 131,0 799 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 100,0 800 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 207,0 801 /home/dcuser/dc_workshop/results/bam/SRR2589044.aligned.sorted.bam 255,0 gt_GT gt_GT_alleles 796 1 T 797 1 G 798 1 AGG 799 1 AC 800 1 G 801 1 Tk.
variants$sample_id[1] "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863" "SRR2584863" [6] "SRR2584863"l.
variants[variants$REF == "A",]sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP 11 SRR2584863 CP000819.1 2407766 NA A C 104 NA FALSE NA NA 9 12 SRR2584863 CP000819.1 2446984 NA A C 225 NA FALSE NA NA 20 14 SRR2584863 CP000819.1 2665639 NA A T 225 NA FALSE NA NA 19 16 SRR2584863 CP000819.1 3339313 NA A C 211 NA FALSE NA NA 10 18 SRR2584863 CP000819.1 3481820 NA A G 200 NA FALSE NA NA 9 19 SRR2584863 CP000819.1 3488669 NA A C 225 NA FALSE NA NA 13 VDB RPB MQB BQB MQSB SGB MQ0F ICB HOB AC 11 0.0230738 0.900802 0.150134 0.750668 0.500000 -0.590765 0.333333 NA NA 1 12 0.0714027 NA NA NA 1.000000 -0.689466 0.000000 NA NA 1 14 0.9960390 NA NA NA 1.000000 -0.690438 0.000000 NA NA 1 16 0.4059360 NA NA NA 1.007750 -0.670168 0.000000 NA NA 1 18 0.1070810 NA NA NA 0.974597 -0.662043 0.000000 NA NA 1 19 0.0162706 NA NA NA 1.000000 -0.680642 0.000000 NA NA 1 AN DP4 MQ 11 1 3,0,3,2 25 12 1 0,0,10,6 60 14 1 0,0,12,5 60 16 1 0,0,4,6 60 18 1 0,0,4,5 60 19 1 0,0,8,4 60 Indiv gt_PL 11 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 131,0 12 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 255,0 14 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 255,0 16 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 241,0 18 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 230,0 19 /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam 255,0 gt_GT gt_GT_alleles 11 1 C 12 1 C 14 1 T 16 1 C 18 1 G 19 1 C
The subsetting notation is very similar to what we learned for vectors. The key differences include:
- Typically provide two values separated by commas: data.frame[row, column]
- In cases where you are taking a continuous range of numbers use a colon between the numbers (start:stop, inclusive)
- For a non continuous set of numbers, pass a vector using
c() - Index using the name of a column(s) by passing them as vectors using
c()
Finally, in all of the subsetting exercises above, we printed values to the screen. You can create a new data frame object by assigning them to a new object name:
# create a new data frame containing only observations from SRR2584863
SRR2584863_variants <- variants[variants$sample_id == "SRR2584863",]
# check the dimension of the data frame
dim(SRR2584863_variants)
[1] 25 29
# get a summary of the data frame
summary(SRR2584863_variants)
sample_id CHROM POS ID
Length:25 Length:25 Min. : 9972 Mode:logical
Class :character Class :character 1st Qu.:1331794 NA's:25
Mode :character Mode :character Median :2618472
Mean :2464989
3rd Qu.:3488669
Max. :4616538
REF ALT QUAL FILTER
Length:25 Length:25 Min. : 31.89 Mode:logical
Class :character Class :character 1st Qu.:104.00 NA's:25
Mode :character Mode :character Median :211.00
Mean :172.97
3rd Qu.:225.00
Max. :228.00
INDEL IDV IMF DP
Mode :logical Min. : 2.00 Min. :0.6667 Min. : 2.0
FALSE:19 1st Qu.: 3.25 1st Qu.:0.9250 1st Qu.: 9.0
TRUE :6 Median : 8.00 Median :1.0000 Median :10.0
Mean : 7.00 Mean :0.9278 Mean :10.4
3rd Qu.: 9.75 3rd Qu.:1.0000 3rd Qu.:12.0
Max. :12.00 Max. :1.0000 Max. :20.0
NA's :19 NA's :19
VDB RPB MQB BQB
Min. :0.01627 Min. :0.9008 Min. :0.04979 Min. :0.7507
1st Qu.:0.07140 1st Qu.:0.9275 1st Qu.:0.09996 1st Qu.:0.7627
Median :0.37674 Median :0.9542 Median :0.15013 Median :0.7748
Mean :0.40429 Mean :0.9517 Mean :0.39997 Mean :0.8418
3rd Qu.:0.65951 3rd Qu.:0.9771 3rd Qu.:0.57507 3rd Qu.:0.8874
Max. :0.99604 Max. :1.0000 Max. :1.00000 Max. :1.0000
NA's :22 NA's :22 NA's :22
MQSB SGB MQ0F ICB
Min. :0.5000 Min. :-0.6904 Min. :0.00000 Mode:logical
1st Qu.:0.9599 1st Qu.:-0.6762 1st Qu.:0.00000 NA's:25
Median :0.9962 Median :-0.6620 Median :0.00000
Mean :0.9442 Mean :-0.6341 Mean :0.04667
3rd Qu.:1.0000 3rd Qu.:-0.6168 3rd Qu.:0.00000
Max. :1.0128 Max. :-0.4536 Max. :0.66667
NA's :3
HOB AC AN DP4 MQ
Mode:logical Min. :1 Min. :1 Length:25 Min. :10.00
NA's:25 1st Qu.:1 1st Qu.:1 Class :character 1st Qu.:60.00
Median :1 Median :1 Mode :character Median :60.00
Mean :1 Mean :1 Mean :55.52
3rd Qu.:1 3rd Qu.:1 3rd Qu.:60.00
Max. :1 Max. :1 Max. :60.00
Indiv gt_PL gt_GT gt_GT_alleles
Length:25 Length:25 Min. :1 Length:25
Class :character Class :character 1st Qu.:1 Class :character
Mode :character Mode :character Median :1 Mode :character
Mean :1
3rd Qu.:1
Max. :1
Coercing values in data frames
Tip: coercion isn’t limited to data frames
While we are going to address coercion in the context of data frames most of these methods apply to other data structures, such as vectors
Sometimes, it is possible that R will misinterpret the type of data represented in a data frame, or store that data in a mode which prevents you from operating on the data the way you wish. For example, a long list of gene names isn’t usually thought of as a categorical variable, the way that your experimental condition (e.g. control, treatment) might be. More importantly, some R packages you use to analyze your data may expect characters as input, not factors. At other times (such as plotting or some statistical analyses) a factor may be more appropriate. Ultimately, you should know how to change the mode of an object.
First, its very important to recognize that coercion happens in R all the time. This can be a good thing when R gets it right, or a bad thing when the result is not what you expect. Consider:
snp_chromosomes <- c('3', '11', 'X', '6')
typeof(snp_chromosomes)
[1] "character"
Although there are several numbers in our vector, they are all in quotes, so we have explicitly told R to consider them as characters. However, even if we removed the quotes from the numbers, R would coerce everything into a character:
snp_chromosomes_2 <- c(3, 11, 'X', 6)
typeof(snp_chromosomes_2)
[1] "character"
snp_chromosomes_2[1]
[1] "3"
We can use the as. functions to explicitly coerce values from one form into
another. Consider the following vector of characters, which all happen to be
valid numbers:
snp_positions_2 <- c("8762685", "66560624", "67545785", "154039662")
typeof(snp_positions_2)
[1] "character"
snp_positions_2[1]
[1] "8762685"
Now we can coerce snp_positions_2 into a numeric type using as.numeric():
snp_positions_2 <- as.numeric(snp_positions_2)
typeof(snp_positions_2)
[1] "double"
snp_positions_2[1]
[1] 8762685
Sometimes coercion is straight forward, but what would happen if we tried
using as.numeric() on snp_chromosomes_2
snp_chromosomes_2 <- as.numeric(snp_chromosomes_2)
Warning: NAs introduced by coercion
If we check, we will see that an NA value (R’s default value for missing
data) has been introduced.
snp_chromosomes_2
[1] 3 11 NA 6
Trouble can really start when we try to coerce a factor. For example, when we
try to coerce the sample_id column in our data frame into a numeric mode
look at the result:
as.numeric(variants$sample_id)
Warning: NAs introduced by coercion
[1] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[26] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[51] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[76] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[101] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[126] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[151] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[176] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[201] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[226] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[251] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[276] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[301] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[326] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[351] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[376] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[401] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[426] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[451] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[476] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[501] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[526] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[551] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[576] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[601] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[626] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[651] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[676] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[701] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[726] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[751] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[776] NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA NA
[801] NA
Strangely, it works! Almost. Instead of giving an error message, R returns numeric values, which in this case are the integers assigned to the levels in this factor. This kind of behavior can lead to hard-to-find bugs, for example when we do have numbers in a factor, and we get numbers from a coercion. If we don’t look carefully, we may not notice a problem.
If you need to coerce an entire column you can overwrite it using an expression like this one:
# make the 'REF' column a character type column
variants$REF <- as.character(variants$REF)
# check the type of the column
typeof(variants$REF)
[1] "character"
StringsAsFactors = ?
Lets summarize this section on coercion with a few take home messages.
- When you explicitly coerce one data type into another (this is known as explicit coercion), be careful to check the result. Ideally, you should try to see if its possible to avoid steps in your analysis that force you to coerce.
- R will sometimes coerce without you asking for it. This is called (appropriately) implicit coercion. For example when we tried to create a vector with multiple data types, R chose one type through implicit coercion.
- Check the structure (
str()) of your data frames before working with them!
Tip: coercion isn’t limited to data frames
Prior to R 4.0 when importing a data frame using any one of the
read.table()functions such asread.csv(), the argumentStringsAsFactorswas by default set to true TRUE. Setting it to FALSE will treat any non-numeric column to a character type.read.csv()documentation, you will also see you can explicitly type your columns using thecolClassesargument. Other R packages (such as the Tidyverse “readr”) don’t have this particular conversion issue, but many packages will still try to guess a data type.
Data frame bonus material: math, sorting, renaming
Here are a few operations that don’t need much explanation, but which are good to know.
There are lots of arithmetic functions you may want to apply to your data frame, covering those would be a course in itself (there is some starting material here). Our lessons will cover some additional summary statistical functions in a subsequent lesson, but overall we will focus on data cleaning and visualization.
You can use functions like mean(), min(), max() on an
individual column. Let’s look at the “DP” or filtered depth. This value shows the number of filtered
reads that support each of the reported variants.
max(variants$DP)
[1] 79
You can sort a data frame using the order() function:
sorted_by_DP <- variants[order(variants$DP), ]
head(sorted_by_DP$DP)
[1] 2 2 2 2 2 2
Exercise
The
order()function lists values in increasing order by default. Look at the documentation for this function and changesorted_by_DPto start with variants with the greatest filtered depth (“DP”).Solution
sorted_by_DP <- variants[order(variants$DP, decreasing = TRUE), ] head(sorted_by_DP$DP)[1] 79 46 41 29 29 27
You can rename columns:
colnames(variants)[colnames(variants) == "sample_id"] <- "strain"
# check the column name (hint names are returned as a vector)
colnames(variants)
[1] "strain" "CHROM" "POS" "ID"
[5] "REF" "ALT" "QUAL" "FILTER"
[9] "INDEL" "IDV" "IMF" "DP"
[13] "VDB" "RPB" "MQB" "BQB"
[17] "MQSB" "SGB" "MQ0F" "ICB"
[21] "HOB" "AC" "AN" "DP4"
[25] "MQ" "Indiv" "gt_PL" "gt_GT"
[29] "gt_GT_alleles"
Saving your data frame to a file
We can save data to a file. We will save our SRR2584863_variants object
to a .csv file using the write.csv() function:
write.csv(SRR2584863_variants, file = "../data/SRR2584863_variants.csv")
The write.csv() function has some additional arguments listed in the help, but
at a minimum you need to tell it what data frame to write to file, and give a
path to a file name in quotes (if you only provide a file name, the file will
be written in the current working directory).
Importing data from Excel
Excel is one of the most common formats, so we need to discuss how to make these files play nicely with R. The simplest way to import data from Excel is to save your Excel file in .csv format*. You can then import into R right away. Sometimes you may not be able to do this (imagine you have data in 300 Excel files, are you going to open and export all of them?).
One common R package (a set of code with features you can download and add to your R installation) is the readxl package which can open and import Excel files. Rather than addressing package installation this second (we’ll discuss this soon!), we can take advantage of RStudio’s import feature which integrates this package. (Note: this feature is available only in the latest versions of RStudio such as is installed on our cloud instance).
First, in the RStudio menu go to File, select Import Dataset, and choose From Excel… (notice there are several other options you can explore).
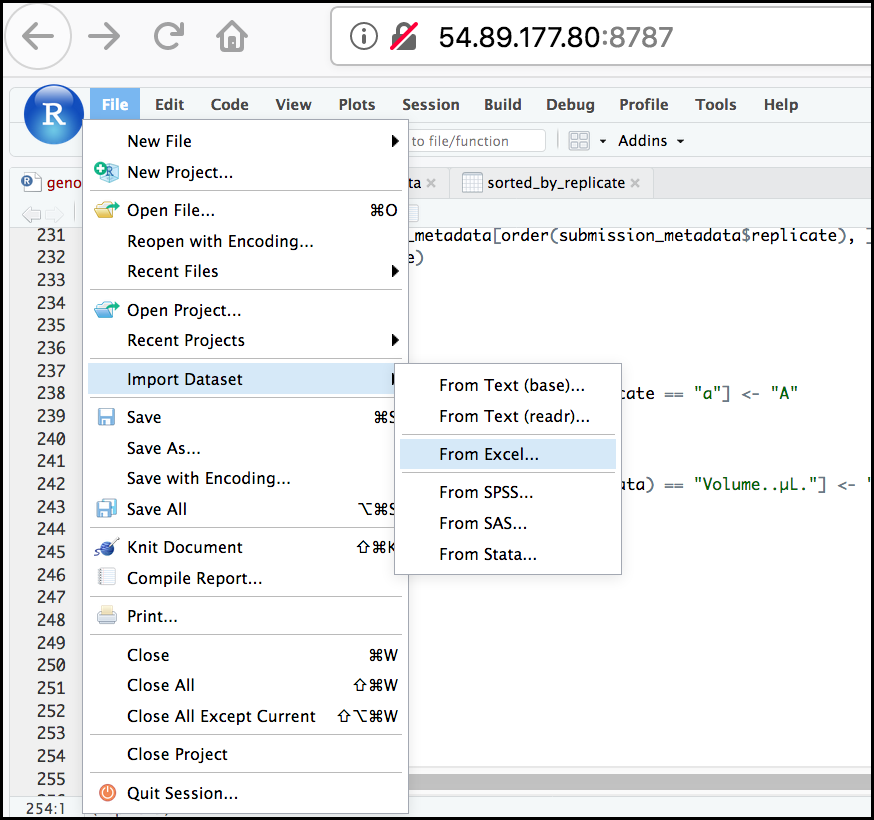
Next, under File/Url: click the Browse button and navigate to the Ecoli_metadata.xlsx file located at /home/dcuser/dc_sample_data/R.
You should now see a preview of the data to be imported:
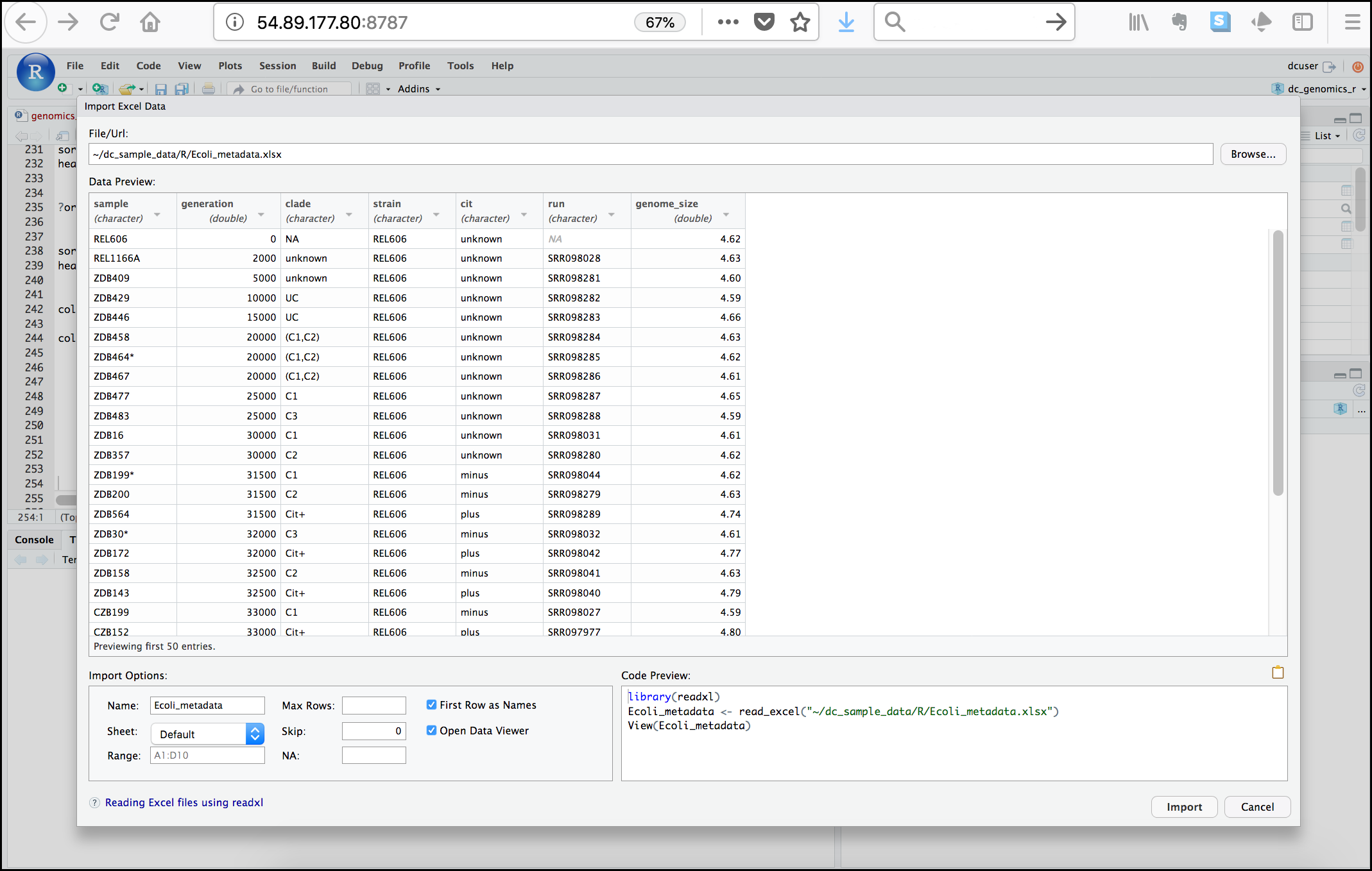
Notice that you have the option to change the data type of each variable by clicking arrow (drop-down menu) next to each column title. Under Import Options you may also rename the data, choose a different sheet to import, and choose how you will handle headers and skipped rows. Under Code Preview you can see the code that will be used to import this file. We could have written this code and imported the Excel file without the RStudio import function, but now you can choose your preference.
In this exercise, we will leave the title of the data frame as Ecoli_metadata, and there are no other options we need to adjust. Click the Import button to import the data.
Finally, let’s check the first few lines of the Ecoli_metadata data
frame:
head(Ecoli_metadata)
# A tibble: 6 × 7
sample generation clade strain cit run genome_size
<chr> <dbl> <chr> <chr> <chr> <chr> <dbl>
1 REL606 0 NA REL606 unknown <NA> 4.62
2 REL1166A 2000 unknown REL606 unknown SRR098028 4.63
3 ZDB409 5000 unknown REL606 unknown SRR098281 4.6
4 ZDB429 10000 UC REL606 unknown SRR098282 4.59
5 ZDB446 15000 UC REL606 unknown SRR098283 4.66
6 ZDB458 20000 (C1,C2) REL606 unknown SRR098284 4.63
The type of this object is ‘tibble’, a type of data
frame we will talk more about in the ‘dplyr’ section. If you needed
a true R data frame you could coerce with as.data.frame().
Exercise: Putting it all together - data frames
Using the
Ecoli_metadatadata frame created above, answer the following questionsA) What are the dimensions (# rows, # columns) of the data frame?
B) What are categories are there in the
citcolumn? hint: treat column as factorC) How many of each of the
citcategories are there?D) What is the genome size for the 7th observation in this data set?
E) What is the median value of the variable
genome_sizeF) Rename the column
sampletosample_idG) Create a new column (name genome_size_bp) and set it equal to the genome_size multiplied by 1,000,000
H) Save the edited Ecoli_metadata data frame as “exercise_solution.csv” in your current working directory.
Solution
dim(Ecoli_metadata)[1] 30 7levels(as.factor(Ecoli_metadata$cit))[1] "minus" "plus" "unknown"table(as.factor(Ecoli_metadata$cit))minus plus unknown 9 9 12Ecoli_metadata[7,7]# A tibble: 1 × 1 genome_size <dbl> 1 4.62median(Ecoli_metadata$genome_size)[1] 4.625colnames(Ecoli_metadata)[colnames(Ecoli_metadata) == "sample"] <- "sample_id" Ecoli_metadata$genome_size_bp <- Ecoli_metadata$genome_size * 1000000 write.csv(Ecoli_metadata, file = "exercise_solution.csv")
Key Points
It is easy to import data into R from tabular formats including Excel. However, you still need to check that R has imported and interpreted your data correctly
There are best practices for organizing your data (keeping it tidy) and R is great for this
Base R has many useful functions for manipulating your data, but all of R’s capabilities are greatly enhanced by software packages developed by the community
Using packages from Bioconductor
Overview
Teaching: 10 min
Exercises: 3 minQuestions
How do I use packages from the Bioconductor repository?
Objectives
Describe what the Bioconductor repository is and what it is used for
Describe how Bioconductor differs from CRAN
Search Bioconductor for relevent packages
Install a package from Bioconductor
Installing packages from somewhere else besides CRAN?
In some cases, you may want to use a specialized package that is not hosted on CRAN (the Comprehensive R Archive Network). This may be because the package is so new that it hasn’t yet been submitted to CRAN, or it could be that it is on a focal topic that has an alternative repository. One major example of an alternative repository source is Bioconductor, which has a mission of “promot[ing] the statistical analysis and comprehension of current and emerging high-throughput biological assays.” This means that many if not all of the packages available on Bioconductor are focused on the analysis of biological data, and that it can be a great place to look for tools to help you analyze your -omics datasets!
So how do I use it?
Since access to the Bioconductor repository is not built in to base R ‘out of the box’, there are a couple steps needed to install packages from this alternative source. We will work through the steps (only 2!) to install a package to help with the VCF analysis we are working on, but you can use the same approach to install any of the many thousands of available packages.

First, install the BiocManager package
The first step is to install a package that is on CRAN, BiocManager. This package will allow us to use it to install packages from Bioconductor. You can think of Bioconductor kind of like an alternative app store for your phone, except instead of apps you are installing packages, and instead of your phone it’s your local R package library.
# install the BiocManager from CRAN using the base R install.packages() function
install.packages("BiocManager")
To check if this worked (and also so you can make a note of the version for reproducibility purposes), you can run BiocManager::version() and it should give you the version number.
# to make sure it worked, check the version
BiocManager::version()
Second, install the vcfR package from Bioconductor using BiocManager
# install the vcfR package from bioconductor using BiocManager::install()
BiocManager::install("vcfR")
You may need to also allow it to install some dependencies or update installed packages in order to successfully complete the process.
Note: Installing packages from Bioconductor vs from CRAN
Some packages begin by being available only on Bioconductor, and then later move to CRAN.
vcfRis one such package, which originally was only available from Bioconductor, but is currently available from CRAN. The other thing to know is thatBiocManager::install()will also install packages from CRAN (it is a wrapper aroundinstall.packages()that adds some extra features). There are other benefits to usingBiocManager::install()for Bioconductor packages, many of which are outlined here. In short, Bioconductor packages have a release cycle that is different from CRAN and theinstall()function is aware of that difference, so it helps to keep package versions in line with one another in a way that doesn’t generally happen with the base Rinstall.packages().
Search for Bioconductor packages based on your analysis needs
While we are only focusing in this workshop on VCF analyses, there are hundreds or thousands of different types of data and analyses that bioinformaticians may want to work with. Sometimes you may get a new dataset and not know exactly where to start with analyzing or visualizing it. The Bioconductor package search view can be a great way to browse through the packages that are available.
Tip: Searching for packages on the Bioconductor website
There are several thousand packages available through the Bioconductor website. It can be a bit of a challenge to find what you want, but one helpful resource is the package search page.
In bioinformatics, there are often many different tools that can be used in a
particular instance. The authors of vcfR have compiled some of
them. One of
those packages that is available from
Bioconductor
is called VariantAnnotation and may also be of interest to those working with
vcf files in R.
Challenge
Add code chunks to
- Install the
BiocManagerpackage- Use that package’s
install()function to installvcfR- Browse the Bioconductor website to find a second package, and install it
Resources
Key Points
Bioconductor is an alternative package repository for bioinformatics packages.
Installing packages from Bioconductor requires a new method, since it is not compatible with the
install.packages()function used for CRAN.Check Bioconductor to see if there is a package relevent to your analysis before writing code yourself.
Data Wrangling and Analyses with Tidyverse
Overview
Teaching: 40 min
Exercises: 15 minQuestions
How can I manipulate data frames without repeating myself?
Objectives
Describe what the
dplyrpackage in R is used for.Apply common
dplyrfunctions to manipulate data in R.Employ the ‘pipe’ operator to link together a sequence of functions.
Employ the ‘mutate’ function to apply other chosen functions to existing columns and create new columns of data.
Employ the ‘split-apply-combine’ concept to split the data into groups, apply analysis to each group, and combine the results.
Bracket subsetting is handy, but it can be cumbersome and difficult to read, especially for complicated operations.
Luckily, the dplyr
package provides a number of very useful functions for manipulating data frames
in a way that will reduce repetition, reduce the probability of making
errors, and probably even save you some typing. As an added bonus, you might
even find the dplyr grammar easier to read.
Here we’re going to cover some of the most commonly used functions as well as using
pipes (%>%) to combine them:
glimpse()select()filter()group_by()summarize()mutate()pivot_longerandpivot_wider
Packages in R are sets of additional functions that let you do more
stuff in R. The functions we’ve been using, like str(), come built into R;
packages give you access to more functions. You need to install a package and
then load it to be able to use it.
install.packages("dplyr") ## installs dplyr package
install.packages("readr") ## install readr pacakge
You might get asked to choose a CRAN mirror – this is asking you to choose a site to download the package from. The choice doesn’t matter too much; I’d recommend choosing the RStudio mirror.
library("dplyr") ## loads in dplyr package to use
library("readr") ## load in readr package to use
You only need to install a package once per computer, but you need to load it every time you open a new R session and want to use that package.
What is dplyr?
The package dplyr is a fairly new (2014) package that tries to provide easy
tools for the most common data manipulation tasks. This package is also included in the tidyverse package, which is a collection of eight different packages (dplyr, ggplot2, tibble, tidyr, readr, purrr, stringr, and forcats). It is built to work directly
with data frames. The thinking behind it was largely inspired by the package
plyr which has been in use for some time but suffered from being slow in some
cases. dplyr addresses this by porting much of the computation to C++. An
additional feature is the ability to work with data stored directly in an
external database. The benefits of doing this are that the data can be managed
natively in a relational database, queries can be conducted on that database,
and only the results of the query returned.
This addresses a common problem with R in that all operations are conducted in memory and thus the amount of data you can work with is limited by available memory. The database connections essentially remove that limitation in that you can have a database that is over 100s of GB, conduct queries on it directly and pull back just what you need for analysis in R.
Loading .csv files in tidy style
The Tidyverse’s readr package provides its own unique way of loading .csv files in to R using read_csv(), which is similar to read.csv(). read_csv() allows users to load in their data faster, doesn’t create row names, and allows you to access non-standard variable names (ie. variables that start with numbers of contain spaces), and outputs your data on the R console in a tidier way. In short, it’s a much friendlier way of loading in potentially messy data.
Now let’s load our vcf .csv file using read_csv():
Taking a quick look at data frames
Similar to str(), which comes built into R, glimpse() is a dplyr function that (as the name suggests) gives a glimpse of the data frame.
Rows: 801
Columns: 29
$ sample_id <chr> "SRR2584863", "SRR2584863", "SRR2584863", "SRR2584863", …
$ CHROM <chr> "CP000819.1", "CP000819.1", "CP000819.1", "CP000819.1", …
$ POS <dbl> 9972, 263235, 281923, 433359, 473901, 648692, 1331794, 1…
$ ID <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ REF <chr> "T", "G", "G", "CTTTTTTT", "CCGC", "C", "C", "G", "ACAGC…
$ ALT <chr> "G", "T", "T", "CTTTTTTTT", "CCGCGC", "T", "A", "A", "AC…
$ QUAL <dbl> 91.0000, 85.0000, 217.0000, 64.0000, 228.0000, 210.0000,…
$ FILTER <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ INDEL <lgl> FALSE, FALSE, FALSE, TRUE, TRUE, FALSE, FALSE, FALSE, TR…
$ IDV <dbl> NA, NA, NA, 12, 9, NA, NA, NA, 2, 7, NA, NA, NA, NA, NA,…
$ IMF <dbl> NA, NA, NA, 1.000000, 0.900000, NA, NA, NA, 0.666667, 1.…
$ DP <dbl> 4, 6, 10, 12, 10, 10, 8, 11, 3, 7, 9, 20, 12, 19, 15, 10…
$ VDB <dbl> 0.0257451, 0.0961330, 0.7740830, 0.4777040, 0.6595050, 0…
$ RPB <dbl> NA, 1.000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.900802, …
$ MQB <dbl> NA, 1.0000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.1501340…
$ BQB <dbl> NA, 1.000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.750668, …
$ MQSB <dbl> NA, NA, 0.974597, 1.000000, 0.916482, 0.916482, 0.900802…
$ SGB <dbl> -0.556411, -0.590765, -0.662043, -0.676189, -0.662043, -…
$ MQ0F <dbl> 0.000000, 0.166667, 0.000000, 0.000000, 0.000000, 0.0000…
$ ICB <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ HOB <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ AC <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ AN <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ DP4 <chr> "0,0,0,4", "0,1,0,5", "0,0,4,5", "0,1,3,8", "1,0,2,7", "…
$ MQ <dbl> 60, 33, 60, 60, 60, 60, 60, 60, 60, 60, 25, 60, 10, 60, …
$ Indiv <chr> "/home/dcuser/dc_workshop/results/bam/SRR2584863.aligned…
$ gt_PL <dbl> 1210, 1120, 2470, 910, 2550, 2400, 2080, 2550, 11128, 19…
$ gt_GT <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ gt_GT_alleles <chr> "G", "T", "T", "CTTTTTTTT", "CCGCGC", "T", "A", "A", "AC…
In the above output, we can already gather some information about variants, such as the number of rows and columns, column names, type of vector in the columns, and the first few entries of each column. Although what we see is similar to outputs of str(), this method gives a cleaner visual output.
Selecting columns and filtering rows
To select columns of a data frame, use select(). The first argument to this function is the data frame (variants), and the subsequent arguments are the columns to keep.
select(variants, sample_id, REF, ALT, DP)
# A tibble: 801 × 4
sample_id REF ALT DP
<chr> <chr> <chr> <dbl>
1 SRR2584863 T G 4
2 SRR2584863 G T 6
3 SRR2584863 G T 10
4 SRR2584863 CTTTTTTT CTTTTTTTT 12
5 SRR2584863 CCGC CCGCGC 10
6 SRR2584863 C T 10
7 SRR2584863 C A 8
8 SRR2584863 G A 11
9 SRR2584863 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG ACAGCCAGCCAGCCAGCCAGCCAGCC… 3
10 SRR2584863 AT ATT 7
# … with 791 more rows
To select all columns except certain ones, put a “-“ in front of the variable to exclude it.
select(variants, -CHROM)
# A tibble: 801 × 28
sampl…¹ POS ID REF ALT QUAL FILTER INDEL IDV IMF DP VDB
<chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl> <dbl>
1 SRR258… 9.97e3 NA T G 91 NA FALSE NA NA 4 0.0257
2 SRR258… 2.63e5 NA G T 85 NA FALSE NA NA 6 0.0961
3 SRR258… 2.82e5 NA G T 217 NA FALSE NA NA 10 0.774
4 SRR258… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12 0.478
5 SRR258… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10 0.660
6 SRR258… 6.49e5 NA C T 210 NA FALSE NA NA 10 0.268
7 SRR258… 1.33e6 NA C A 178 NA FALSE NA NA 8 0.624
8 SRR258… 1.73e6 NA G A 225 NA FALSE NA NA 11 0.992
9 SRR258… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3 0.902
10 SRR258… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7 0.568
# … with 791 more rows, 16 more variables: RPB <dbl>, MQB <dbl>, BQB <dbl>,
# MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>, AC <dbl>,
# AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>, gt_GT <dbl>,
# gt_GT_alleles <chr>, and abbreviated variable name ¹sample_id
dplyr also provides useful functions to select columns based on their names. For instance, ends_with() allows you to select columns that ends with specific letters. For instance, if you wanted to select columns that end with the letter “B”:
select(variants, ends_with("B"))
# A tibble: 801 × 8
VDB RPB MQB BQB MQSB SGB ICB HOB
<dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <lgl> <lgl>
1 0.0257 NA NA NA NA -0.556 NA NA
2 0.0961 1 1 1 NA -0.591 NA NA
3 0.774 NA NA NA 0.975 -0.662 NA NA
4 0.478 NA NA NA 1 -0.676 NA NA
5 0.660 NA NA NA 0.916 -0.662 NA NA
6 0.268 NA NA NA 0.916 -0.670 NA NA
7 0.624 NA NA NA 0.901 -0.651 NA NA
8 0.992 NA NA NA 1.01 -0.670 NA NA
9 0.902 NA NA NA 1 -0.454 NA NA
10 0.568 NA NA NA 1.01 -0.617 NA NA
# … with 791 more rows
Challenge
Create a table that contains all the columns with the letter “i” and column “POS”, without columns “Indiv” and “FILTER”. Hint: look at the help function
ends_with()we’ve just covered.Solution
select(variants, contains("i"), -Indiv, -FILTER, POS)# A tibble: 801 × 7 sample_id ID INDEL IDV IMF ICB POS <chr> <lgl> <lgl> <dbl> <dbl> <lgl> <dbl> 1 SRR2584863 NA FALSE NA NA NA 9972 2 SRR2584863 NA FALSE NA NA NA 263235 3 SRR2584863 NA FALSE NA NA NA 281923 4 SRR2584863 NA TRUE 12 1 NA 433359 5 SRR2584863 NA TRUE 9 0.9 NA 473901 6 SRR2584863 NA FALSE NA NA NA 648692 7 SRR2584863 NA FALSE NA NA NA 1331794 8 SRR2584863 NA FALSE NA NA NA 1733343 9 SRR2584863 NA TRUE 2 0.667 NA 2103887 10 SRR2584863 NA TRUE 7 1 NA 2333538 # … with 791 more rows
To choose rows, use filter():
filter(variants, sample_id == "SRR2584863")
# A tibble: 25 × 29
sample…¹ CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR2584… CP00… 9.97e3 NA T G 91 NA FALSE NA NA 4
2 SRR2584… CP00… 2.63e5 NA G T 85 NA FALSE NA NA 6
3 SRR2584… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
4 SRR2584… CP00… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12
5 SRR2584… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
6 SRR2584… CP00… 6.49e5 NA C T 210 NA FALSE NA NA 10
7 SRR2584… CP00… 1.33e6 NA C A 178 NA FALSE NA NA 8
8 SRR2584… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
9 SRR2584… CP00… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3
10 SRR2584… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
# … with 15 more rows, 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>, and abbreviated variable name ¹sample_id
filter() will keep all the rows that match the conditions that are provided. Here are a few examples:
# rows for which the reference genome has T or G
filter(variants, REF %in% c("T", "G"))
# A tibble: 340 × 29
sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR25848… CP00… 9.97e3 NA T G 91 NA FALSE NA NA 4
2 SRR25848… CP00… 2.63e5 NA G T 85 NA FALSE NA NA 6
3 SRR25848… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
4 SRR25848… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
5 SRR25848… CP00… 2.62e6 NA G T 31.9 NA FALSE NA NA 12
6 SRR25848… CP00… 3.00e6 NA G A 225 NA FALSE NA NA 15
7 SRR25848… CP00… 3.91e6 NA G T 225 NA FALSE NA NA 10
8 SRR25848… CP00… 9.97e3 NA T G 214 NA FALSE NA NA 10
9 SRR25848… CP00… 1.06e4 NA G A 225 NA FALSE NA NA 11
10 SRR25848… CP00… 6.40e4 NA G A 225 NA FALSE NA NA 18
# … with 330 more rows, and 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>
# rows with QUAL values greater than or equal to 100
filter(variants, QUAL >= 100)
# A tibble: 666 × 29
sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR25848… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
2 SRR25848… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
3 SRR25848… CP00… 6.49e5 NA C T 210 NA FALSE NA NA 10
4 SRR25848… CP00… 1.33e6 NA C A 178 NA FALSE NA NA 8
5 SRR25848… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
6 SRR25848… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
7 SRR25848… CP00… 2.41e6 NA A C 104 NA FALSE NA NA 9
8 SRR25848… CP00… 2.45e6 NA A C 225 NA FALSE NA NA 20
9 SRR25848… CP00… 2.67e6 NA A T 225 NA FALSE NA NA 19
10 SRR25848… CP00… 3.00e6 NA G A 225 NA FALSE NA NA 15
# … with 656 more rows, and 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>
# rows that have TRUE in the column INDEL
filter(variants, INDEL)
# A tibble: 101 × 29
sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR25848… CP00… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12
2 SRR25848… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
3 SRR25848… CP00… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3
4 SRR25848… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
5 SRR25848… CP00… 3.90e6 NA A AC 43.4 NA TRUE 2 1 2
6 SRR25848… CP00… 4.43e6 NA TGG T 228 NA TRUE 10 1 10
7 SRR25848… CP00… 1.48e5 NA AGGGG AGGG… 122 NA TRUE 8 1 8
8 SRR25848… CP00… 1.58e5 NA GTTT… GTTT… 19.5 NA TRUE 6 1 6
9 SRR25848… CP00… 1.73e5 NA CAA CA 180 NA TRUE 11 1 11
10 SRR25848… CP00… 1.75e5 NA GAA GA 194 NA TRUE 10 1 10
# … with 91 more rows, and 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>
# rows that don't have missing data in the IDV column
filter(variants, !is.na(IDV))
# A tibble: 101 × 29
sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR25848… CP00… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12
2 SRR25848… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
3 SRR25848… CP00… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3
4 SRR25848… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
5 SRR25848… CP00… 3.90e6 NA A AC 43.4 NA TRUE 2 1 2
6 SRR25848… CP00… 4.43e6 NA TGG T 228 NA TRUE 10 1 10
7 SRR25848… CP00… 1.48e5 NA AGGGG AGGG… 122 NA TRUE 8 1 8
8 SRR25848… CP00… 1.58e5 NA GTTT… GTTT… 19.5 NA TRUE 6 1 6
9 SRR25848… CP00… 1.73e5 NA CAA CA 180 NA TRUE 11 1 11
10 SRR25848… CP00… 1.75e5 NA GAA GA 194 NA TRUE 10 1 10
# … with 91 more rows, and 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>
filter() allows you to combine multiple conditions. You can separate them using a , as arguments to the function, they will be combined using the & (AND) logical operator. If you need to use the | (OR) logical operator, you can specify it explicitly:
# this is equivalent to:
# filter(variants, sample_id == "SRR2584863" & QUAL >= 100)
filter(variants, sample_id == "SRR2584863", QUAL >= 100)
# A tibble: 19 × 29
sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR25848… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
2 SRR25848… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
3 SRR25848… CP00… 6.49e5 NA C T 210 NA FALSE NA NA 10
4 SRR25848… CP00… 1.33e6 NA C A 178 NA FALSE NA NA 8
5 SRR25848… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
6 SRR25848… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
7 SRR25848… CP00… 2.41e6 NA A C 104 NA FALSE NA NA 9
8 SRR25848… CP00… 2.45e6 NA A C 225 NA FALSE NA NA 20
9 SRR25848… CP00… 2.67e6 NA A T 225 NA FALSE NA NA 19
10 SRR25848… CP00… 3.00e6 NA G A 225 NA FALSE NA NA 15
11 SRR25848… CP00… 3.34e6 NA A C 211 NA FALSE NA NA 10
12 SRR25848… CP00… 3.40e6 NA C A 225 NA FALSE NA NA 14
13 SRR25848… CP00… 3.48e6 NA A G 200 NA FALSE NA NA 9
14 SRR25848… CP00… 3.49e6 NA A C 225 NA FALSE NA NA 13
15 SRR25848… CP00… 3.91e6 NA G T 225 NA FALSE NA NA 10
16 SRR25848… CP00… 4.10e6 NA A G 225 NA FALSE NA NA 16
17 SRR25848… CP00… 4.20e6 NA A C 225 NA FALSE NA NA 11
18 SRR25848… CP00… 4.43e6 NA TGG T 228 NA TRUE 10 1 10
19 SRR25848… CP00… 4.62e6 NA A C 185 NA FALSE NA NA 9
# … with 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>, BQB <dbl>,
# MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>, AC <dbl>,
# AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>, gt_GT <dbl>,
# gt_GT_alleles <chr>
# using `|` logical operator
filter(variants, sample_id == "SRR2584863", (INDEL | QUAL >= 100))
# A tibble: 22 × 29
sample…¹ CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR2584… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
2 SRR2584… CP00… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12
3 SRR2584… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
4 SRR2584… CP00… 6.49e5 NA C T 210 NA FALSE NA NA 10
5 SRR2584… CP00… 1.33e6 NA C A 178 NA FALSE NA NA 8
6 SRR2584… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
7 SRR2584… CP00… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3
8 SRR2584… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
9 SRR2584… CP00… 2.41e6 NA A C 104 NA FALSE NA NA 9
10 SRR2584… CP00… 2.45e6 NA A C 225 NA FALSE NA NA 20
# … with 12 more rows, 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>, and abbreviated variable name ¹sample_id
Challenge
Select all the mutations that occurred between the positions 1e6 (one million) and 2e6 (included) that are not indels and have QUAL greater than 200.
Solution
filter(variants, POS >= 1e6 & POS <= 2e6, !INDEL, QUAL > 200)# A tibble: 77 × 29 sample_id CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP <chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl> 1 SRR25848… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11 2 SRR25848… CP00… 1.00e6 NA A G 225 NA FALSE NA NA 15 3 SRR25848… CP00… 1.02e6 NA A G 225 NA FALSE NA NA 12 4 SRR25848… CP00… 1.06e6 NA C T 225 NA FALSE NA NA 17 5 SRR25848… CP00… 1.06e6 NA A G 206 NA FALSE NA NA 9 6 SRR25848… CP00… 1.07e6 NA G T 225 NA FALSE NA NA 11 7 SRR25848… CP00… 1.07e6 NA T C 225 NA FALSE NA NA 12 8 SRR25848… CP00… 1.10e6 NA C T 225 NA FALSE NA NA 15 9 SRR25848… CP00… 1.11e6 NA C T 212 NA FALSE NA NA 9 10 SRR25848… CP00… 1.11e6 NA A G 225 NA FALSE NA NA 14 # … with 67 more rows, and 17 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>, # BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>, # AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>, # gt_GT <dbl>, gt_GT_alleles <chr>
Pipes
But what if you wanted to select and filter? We can do this with pipes. Pipes, are a fairly recent addition to R. Pipes let you
take the output of one function and send it directly to the next, which is
useful when you need to many things to the same data set. It was
possible to do this before pipes were added to R, but it was
much messier and more difficult. Pipes in R look like
%>% and are made available via the magrittr package, which is installed as
part of dplyr. If you use RStudio, you can type the pipe with
Ctrl + Shift + M if you’re using a PC,
or Cmd + Shift + M if you’re using a Mac.
variants %>%
filter(sample_id == "SRR2584863") %>%
select(REF, ALT, DP)
# A tibble: 25 × 3
REF ALT DP
<chr> <chr> <dbl>
1 T G 4
2 G T 6
3 G T 10
4 CTTTTTTT CTTTTTTTT 12
5 CCGC CCGCGC 10
6 C T 10
7 C A 8
8 G A 11
9 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGC… 3
10 AT ATT 7
# … with 15 more rows
In the above code, we use the pipe to send the variants dataset first through
filter(), to keep rows where sample_id matches a particular sample, and then through select() to
keep only the REF, ALT, and DP columns. Since %>% takes
the object on its left and passes it as the first argument to the function on
its right, we don’t need to explicitly include the data frame as an argument
to the filter() and select() functions any more.
Some may find it helpful to read the pipe like the word “then”. For instance,
in the above example, we took the data frame variants, then we filtered
for rows where sample_id was SRR2584863, then we selected the REF, ALT, and DP columns, then we showed only the first six rows.
The dplyr functions by themselves are somewhat simple,
but by combining them into linear workflows with the pipe, we can accomplish
more complex manipulations of data frames.
If we want to create a new object with this smaller version of the data we can do so by assigning it a new name:
SRR2584863_variants <- variants %>%
filter(sample_id == "SRR2584863") %>%
select(REF, ALT, DP)
This new object includes all of the data from this sample. Let’s look at just the first six rows to confirm it’s what we want:
SRR2584863_variants
# A tibble: 25 × 3
REF ALT DP
<chr> <chr> <dbl>
1 T G 4
2 G T 6
3 G T 10
4 CTTTTTTT CTTTTTTTT 12
5 CCGC CCGCGC 10
6 C T 10
7 C A 8
8 G A 11
9 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGC… 3
10 AT ATT 7
# … with 15 more rows
Similar to head() and tail() functions, we can also look at the first or last six rows using tidyverse function slice(). Slice is a more versatile function that allows users to specify a range to view:
SRR2584863_variants %>% slice(1:6)
# A tibble: 6 × 3
REF ALT DP
<chr> <chr> <dbl>
1 T G 4
2 G T 6
3 G T 10
4 CTTTTTTT CTTTTTTTT 12
5 CCGC CCGCGC 10
6 C T 10
SRR2584863_variants %>% slice(10:25)
# A tibble: 16 × 3
REF ALT DP
<chr> <chr> <dbl>
1 AT ATT 7
2 A C 9
3 A C 20
4 G T 12
5 A T 19
6 G A 15
7 A C 10
8 C A 14
9 A G 9
10 A C 13
11 A AC 2
12 G T 10
13 A G 16
14 A C 11
15 TGG T 10
16 A C 9
Exercise: Pipe and filter
Starting with the
variantsdata frame, use pipes to subset the data to include only observations from SRR2584863 sample, where the filtered depth (DP) is at least 10. Shwoing only 5th through 11th rows of columnsREF,ALT, andPOS.Solution
variants %>% filter(sample_id == "SRR2584863" & DP >= 10) %>% slice(5:11) %>% select(REF, ALT, POS)# A tibble: 7 × 3 REF ALT POS <chr> <chr> <dbl> 1 G A 1733343 2 A C 2446984 3 G T 2618472 4 A T 2665639 5 G A 2999330 6 A C 3339313 7 C A 3401754
Mutate
Frequently you’ll want to create new columns based on the values in existing
columns, for example to do unit conversions or find the ratio of values in two
columns. For this we’ll use the dplyr function mutate().
We have a column titled “QUAL”. This is a Phred-scaled confidence score that a polymorphism exists at this position given the sequencing data. Lower QUAL scores indicate low probability of a polymorphism existing at that site. We can convert the confidence value QUAL to a probability value according to the formula:
Probability = 1- 10 ^ -(QUAL/10)
Let’s add a column (POLPROB) to our variants data frame that shows
the probability of a polymorphism at that site given the data.
variants %>%
mutate(POLPROB = 1 - (10 ^ -(QUAL/10)))
# A tibble: 801 × 30
sample…¹ CHROM POS ID REF ALT QUAL FILTER INDEL IDV IMF DP
<chr> <chr> <dbl> <lgl> <chr> <chr> <dbl> <lgl> <lgl> <dbl> <dbl> <dbl>
1 SRR2584… CP00… 9.97e3 NA T G 91 NA FALSE NA NA 4
2 SRR2584… CP00… 2.63e5 NA G T 85 NA FALSE NA NA 6
3 SRR2584… CP00… 2.82e5 NA G T 217 NA FALSE NA NA 10
4 SRR2584… CP00… 4.33e5 NA CTTT… CTTT… 64 NA TRUE 12 1 12
5 SRR2584… CP00… 4.74e5 NA CCGC CCGC… 228 NA TRUE 9 0.9 10
6 SRR2584… CP00… 6.49e5 NA C T 210 NA FALSE NA NA 10
7 SRR2584… CP00… 1.33e6 NA C A 178 NA FALSE NA NA 8
8 SRR2584… CP00… 1.73e6 NA G A 225 NA FALSE NA NA 11
9 SRR2584… CP00… 2.10e6 NA ACAG… ACAG… 56 NA TRUE 2 0.667 3
10 SRR2584… CP00… 2.33e6 NA AT ATT 167 NA TRUE 7 1 7
# … with 791 more rows, 18 more variables: VDB <dbl>, RPB <dbl>, MQB <dbl>,
# BQB <dbl>, MQSB <dbl>, SGB <dbl>, MQ0F <dbl>, ICB <lgl>, HOB <lgl>,
# AC <dbl>, AN <dbl>, DP4 <chr>, MQ <dbl>, Indiv <chr>, gt_PL <dbl>,
# gt_GT <dbl>, gt_GT_alleles <chr>, POLPROB <dbl>, and abbreviated variable
# name ¹sample_id
Exercise
There are a lot of columns in our dataset, so let’s just look at the
sample_id,POS,QUAL, andPOLPROBcolumns for now. Add a line to the above code to only show those columns.Solution
variants %>% mutate(POLPROB = 1 - 10 ^ -(QUAL/10)) %>% select(sample_id, POS, QUAL, POLPROB)# A tibble: 801 × 4 sample_id POS QUAL POLPROB <chr> <dbl> <dbl> <dbl> 1 SRR2584863 9972 91 1.00 2 SRR2584863 263235 85 1.00 3 SRR2584863 281923 217 1 4 SRR2584863 433359 64 1.00 5 SRR2584863 473901 228 1 6 SRR2584863 648692 210 1 7 SRR2584863 1331794 178 1 8 SRR2584863 1733343 225 1 9 SRR2584863 2103887 56 1.00 10 SRR2584863 2333538 167 1 # … with 791 more rows
group_by() and summarize() functions
Many data analysis tasks can be approached using the “split-apply-combine”
paradigm: split the data into groups, apply some analysis to each group, and
then combine the results. dplyr makes this very easy through the use of the
group_by() function, which splits the data into groups. When the data is
grouped in this way summarize() can be used to collapse each group into
a single-row summary. summarize() does this by applying an aggregating
or summary function to each group. For example, if we wanted to group
by sample_id and find the number of rows of data for each
sample, we would do:
variants %>%
group_by(sample_id) %>%
summarize(n())
# A tibble: 3 × 2
sample_id `n()`
<chr> <int>
1 SRR2584863 25
2 SRR2584866 766
3 SRR2589044 10
It can be a bit tricky at first, but we can imagine physically splitting the data frame by groups and applying a certain function to summarize the data.
^[The figure was adapted from the Software Carpentry lesson, R for Reproducible Scientific Analysis]
Here the summary function used was n() to find the count for each
group. Since this is a quite a common operation, there is a simpler method
called tally():
variants %>%
group_by(ALT) %>%
tally()
# A tibble: 57 × 2
ALT n
<chr> <int>
1 A 211
2 AC 2
3 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 1
4 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 1
5 ACCCCC 2
6 ACCCCCCCC 2
7 AGCGCGCGCG 1
8 AGG 1
9 AGGGGG 2
10 AGGGGGG 2
# … with 47 more rows
To show that there are many ways to achieve the same results, there is another way to approach this, which bypasses group_by() using the function count():
variants %>%
count(ALT)
# A tibble: 57 × 2
ALT n
<chr> <int>
1 A 211
2 AC 2
3 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 1
4 ACAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAGCCAG 1
5 ACCCCC 2
6 ACCCCCCCC 2
7 AGCGCGCGCG 1
8 AGG 1
9 AGGGGG 2
10 AGGGGGG 2
# … with 47 more rows
Challenge
- How many mutations are found in each sample?
Solution
variants %>% count(sample_id)# A tibble: 3 × 2 sample_id n <chr> <int> 1 SRR2584863 25 2 SRR2584866 766 3 SRR2589044 10
We can also apply many other functions to individual columns to get other
summary statistics. For example,we can use built-in functions like mean(),
median(), min(), and max(). These are called “built-in functions” because
they come with R and don’t require that you install any additional packages.
By default, all R functions operating on vectors that contains missing data will return NA.
It’s a way to make sure that users know they have missing data, and make a
conscious decision on how to deal with it. When dealing with simple statistics
like the mean, the easiest way to ignore NA (the missing data) is
to use na.rm = TRUE (rm stands for remove).
So to view the mean, median, maximum, and minimum filtered depth (DP) for each sample:
variants %>%
group_by(sample_id) %>%
summarize(
mean_DP = mean(DP),
median_DP = median(DP),
min_DP = min(DP),
max_DP = max(DP))
# A tibble: 3 × 5
sample_id mean_DP median_DP min_DP max_DP
<chr> <dbl> <dbl> <dbl> <dbl>
1 SRR2584863 10.4 10 2 20
2 SRR2584866 10.6 10 2 79
3 SRR2589044 9.3 9.5 3 16
Reshaping data frames
It can sometimes be useful to transform the “long” tidy format, into the wide format. This transformation can be done with the pivot_wider() function provided by the tidyr package (also part of the tidyverse).
pivot_wider() takes a data frame as the first argument, and two arguments: the column name that will become the columns and the column name that will become the cells in the wide data.
variants_wide <- variants %>%
group_by(sample_id, CHROM) %>%
summarize(mean_DP = mean(DP)) %>%
pivot_wider(names_from = sample_id, values_from = mean_DP)
Error in pivot_wider(., names_from = sample_id, values_from = mean_DP): could not find function "pivot_wider"
variants_wide
Error in eval(expr, envir, enclos): object 'variants_wide' not found
The opposite operation of pivot_wider() is taken care by pivot_longer(). We specify the names of the new columns, and here add -CHROM as this column shouldn’t be affected by the reshaping:
variants_wide %>%
pivot_longer(-CHROM, names_to = "sample_id", values_to = "mean_DP")
Error in pivot_longer(., -CHROM, names_to = "sample_id", values_to = "mean_DP"): could not find function "pivot_longer"
Resources
Key Points
Use the
dplyrpackage to manipulate data frames.Use
glimpse()to quickly look at your data frame.Use
select()to choose variables from a data frame.Use
filter()to choose data based on values.Use
mutate()to create new variables.Use
group_by()andsummarize()to work with subsets of data.
Data Visualization with ggplot2
Overview
Teaching: 60 min
Exercises: 30 minQuestions
What is ggplot2?
What is mapping, and what is aesthetics?
What is the process of creating a publication-quality plots with ggplot in R?
Objectives
Describe the role of data, aesthetics, and geoms in ggplot functions.
Choose the correct aesthetics and alter the geom parameters for a scatter plot, histogram, or box plot.
Layer multiple geometries in a single plot.
Customize plot scales, titles, themes, and fonts.
Apply a facet to a plot.
Apply additional ggplot2-compatible plotting libraries.
Save a ggplot to a file.
List several resources for getting help with ggplot.
List several resources for creating informative scientific plots.
Introduction to ggplot2
![]()
ggplot2 is a plotting package that makes it simple to create complex plots from data in a data frame. It provides a more programmatic interface for specifying what variables to plot, how they are displayed, and general visual properties. Therefore, we only need minimal changes if the underlying data change or if we decide to change from a bar plot to a scatter plot. This helps in creating publication-quality plots with minimal amounts of adjustments and tweaking.
The gg in “ggplot” stands for “Grammar of Graphics,” which is an elegant yet powerful way to describe the making of scientific plots. In short, the grammar of graphics breaks down every plot into a few components, namely, a dataset, a set of geoms (visual marks that represent the data points), and a coordinate system. You can imagine this is a grammar that gives unique names to each component appearing in a plot and conveys specific information about data. With ggplot, graphics are built step by step by adding new elements.
The idea of mapping is crucial in ggplot. One familiar example is to map the value of one variable in a dataset to $x$ and the other to $y$. However, we often encounter datasets that include multiple (more than two) variables. In this case, ggplot allows you to map those other variables to visual marks such as color and shape (aesthetics or aes). One thing you may want to remember is the difference between discrete and continuous variables. Some aesthetics, such as the shape of dots, do not accept continuous variables. If forced to do so, R will give an error. This is easy to understand; we cannot create a continuum of shapes for a variable, unlike, say, color.
Tip: when having doubts about whether a variable is continuous or discrete, a quick way to check is to use the summary() function. Continuous variables have descriptive statistics but not the discrete variables.
Installing tidyverse
ggplot2 belongs to the tidyverse framework. Therefore, we will start with loading the package tidyverse. If tidyverse is not already installed, then we need to install first. If it is already installed, then we can skip the following step:
install.packages("tidyverse") # Installing tidyverse package, includes ggplot2 and other packages such as dplyr, readr, tidyr
Now, let’s load the tidyverse package:
library(tidyverse)
── Attaching packages ─────────────────────────────────────── tidyverse 1.3.2 ──
✔ ggplot2 3.3.6 ✔ purrr 0.3.5
✔ tibble 3.1.8 ✔ dplyr 1.0.10
✔ tidyr 1.2.1 ✔ stringr 1.4.1
✔ readr 2.1.3 ✔ forcats 0.5.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
As we can see from above output ggplot2 has been already loaded along with other packages as part of the tidyverse framework.
Loading the dataset
variants = read_csv("https://raw.githubusercontent.com/naupaka/vcfr-for-data-carpentry-draft/main/output/combined_tidy_vcf.csv")
Rows: 801 Columns: 29
── Column specification ────────────────────────────────────────────────────────
Delimiter: ","
chr (7): sample_id, CHROM, REF, ALT, DP4, Indiv, gt_GT_alleles
dbl (16): POS, QUAL, IDV, IMF, DP, VDB, RPB, MQB, BQB, MQSB, SGB, MQ0F, AC, ...
num (1): gt_PL
lgl (5): ID, FILTER, INDEL, ICB, HOB
ℹ Use `spec()` to retrieve the full column specification for this data.
ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
Explore the structure (types of columns and number of rows) of the dataset using dplyr’s glimpse() (for more info, see the Data Wrangling and Analyses with Tidyverse episode)
glimpse(variants) # Show a snapshot of the rows and columns
Rows: 801
Columns: 29
$ sample_id <chr> "SRR2584863", "SRR2584863", "SRR2584863", "SRR2584863", …
$ CHROM <chr> "CP000819.1", "CP000819.1", "CP000819.1", "CP000819.1", …
$ POS <dbl> 9972, 263235, 281923, 433359, 473901, 648692, 1331794, 1…
$ ID <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ REF <chr> "T", "G", "G", "CTTTTTTT", "CCGC", "C", "C", "G", "ACAGC…
$ ALT <chr> "G", "T", "T", "CTTTTTTTT", "CCGCGC", "T", "A", "A", "AC…
$ QUAL <dbl> 91.0000, 85.0000, 217.0000, 64.0000, 228.0000, 210.0000,…
$ FILTER <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ INDEL <lgl> FALSE, FALSE, FALSE, TRUE, TRUE, FALSE, FALSE, FALSE, TR…
$ IDV <dbl> NA, NA, NA, 12, 9, NA, NA, NA, 2, 7, NA, NA, NA, NA, NA,…
$ IMF <dbl> NA, NA, NA, 1.000000, 0.900000, NA, NA, NA, 0.666667, 1.…
$ DP <dbl> 4, 6, 10, 12, 10, 10, 8, 11, 3, 7, 9, 20, 12, 19, 15, 10…
$ VDB <dbl> 0.0257451, 0.0961330, 0.7740830, 0.4777040, 0.6595050, 0…
$ RPB <dbl> NA, 1.000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.900802, …
$ MQB <dbl> NA, 1.0000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.1501340…
$ BQB <dbl> NA, 1.000000, NA, NA, NA, NA, NA, NA, NA, NA, 0.750668, …
$ MQSB <dbl> NA, NA, 0.974597, 1.000000, 0.916482, 0.916482, 0.900802…
$ SGB <dbl> -0.556411, -0.590765, -0.662043, -0.676189, -0.662043, -…
$ MQ0F <dbl> 0.000000, 0.166667, 0.000000, 0.000000, 0.000000, 0.0000…
$ ICB <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ HOB <lgl> NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, NA, …
$ AC <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ AN <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ DP4 <chr> "0,0,0,4", "0,1,0,5", "0,0,4,5", "0,1,3,8", "1,0,2,7", "…
$ MQ <dbl> 60, 33, 60, 60, 60, 60, 60, 60, 60, 60, 25, 60, 10, 60, …
$ Indiv <chr> "/home/dcuser/dc_workshop/results/bam/SRR2584863.aligned…
$ gt_PL <dbl> 1210, 1120, 2470, 910, 2550, 2400, 2080, 2550, 11128, 19…
$ gt_GT <dbl> 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1, 1,…
$ gt_GT_alleles <chr> "G", "T", "T", "CTTTTTTTT", "CCGCGC", "T", "A", "A", "AC…
Alternatively, we can display the first a few rows (vertically) of the table using head():
head(variants)
| sample_id | CHROM | POS | ID | REF | ALT | QUAL | FILTER | INDEL | IDV | IMF | DP | VDB | RPB | MQB | BQB | MQSB | SGB | MQ0F | ICB | HOB | AC | AN | DP4 | MQ | Indiv | gt_PL | gt_GT | gt_GT_alleles |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SRR2584863 | CP000819.1 | 9972 | NA | T | G | 91 | NA | FALSE | NA | NA | 4 | 0.0257451 | NA | NA | NA | NA | -0.556411 | 0.000000 | NA | NA | 1 | 1 | 0,0,0,4 | 60 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 1210 | 1 | G |
| SRR2584863 | CP000819.1 | 263235 | NA | G | T | 85 | NA | FALSE | NA | NA | 6 | 0.0961330 | 1 | 1 | 1 | NA | -0.590765 | 0.166667 | NA | NA | 1 | 1 | 0,1,0,5 | 33 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 1120 | 1 | T |
| SRR2584863 | CP000819.1 | 281923 | NA | G | T | 217 | NA | FALSE | NA | NA | 10 | 0.7740830 | NA | NA | NA | 0.974597 | -0.662043 | 0.000000 | NA | NA | 1 | 1 | 0,0,4,5 | 60 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 2470 | 1 | T |
| SRR2584863 | CP000819.1 | 433359 | NA | CTTTTTTT | CTTTTTTTT | 64 | NA | TRUE | 12 | 1.0 | 12 | 0.4777040 | NA | NA | NA | 1.000000 | -0.676189 | 0.000000 | NA | NA | 1 | 1 | 0,1,3,8 | 60 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 910 | 1 | CTTTTTTTT |
| SRR2584863 | CP000819.1 | 473901 | NA | CCGC | CCGCGC | 228 | NA | TRUE | 9 | 0.9 | 10 | 0.6595050 | NA | NA | NA | 0.916482 | -0.662043 | 0.000000 | NA | NA | 1 | 1 | 1,0,2,7 | 60 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 2550 | 1 | CCGCGC |
| SRR2584863 | CP000819.1 | 648692 | NA | C | T | 210 | NA | FALSE | NA | NA | 10 | 0.2680140 | NA | NA | NA | 0.916482 | -0.670168 | 0.000000 | NA | NA | 1 | 1 | 0,0,7,3 | 60 | /home/dcuser/dc_workshop/results/bam/SRR2584863.aligned.sorted.bam | 2400 | 1 | T |
ggplot2 functions like data in the long format, i.e., a column for every dimension (variable), and a row for every observation. Well-structured data will save you time when making figures with ggplot2
ggplot2 graphics are built step-by-step by adding new elements. Adding layers in this fashion allows for extensive flexibility and customization of plots, and more equally important the readability of the code.
To build a ggplot, we will use the following basic template that can be used for different types of plots:
ggplot(data = <DATA>, mapping = aes(<MAPPINGS>)) + <GEOM_FUNCTION>()
- use the
ggplot()function and bind the plot to a specific data frame using thedataargument
ggplot(data = variants)
- define a mapping (using the aesthetic (
aes) function), by selecting the variables to be plotted and specifying how to present them in the graph, e.g. as x and y positions or characteristics such as size, shape, color, etc.
ggplot(data = variants, aes(x = POS, y = DP))
- add ‘geoms’ – graphical representations of the data in the plot (points,
lines, bars).
ggplot2offers many different geoms; we will use some common ones today, including:geom_point()for scatter plots, dot plots, etc.geom_boxplot()for, well, boxplots!geom_line()for trend lines, time series, etc.
To add a geom to the plot use the + operator. Because we have two continuous variables, let’s use geom_point() (i.e., a scatter plot) first:
ggplot(data = variants, aes(x = POS, y = DP)) +
geom_point()
The + in the ggplot2 package is particularly useful because it allows you to modify existing ggplot objects. This means you can easily set up plot templates and conveniently explore different types of plots, so the above plot can also be generated with code like this:
# Assign plot to a variable
coverage_plot <- ggplot(data = variants, aes(x = POS, y = DP))
# Draw the plot
coverage_plot +
geom_point()
Notes
- Anything you put in the
ggplot()function can be seen by any geom layers that you add (i.e., these are universal plot settings). This includes the x- and y-axis mapping you set up inaes(). - You can also specify mappings for a given geom independently of the mappings defined globally in the
ggplot()function. - The
+sign used to add new layers must be placed at the end of the line containing the previous layer. If, instead, the+sign is added at the beginning of the line containing the new layer,ggplot2will not add the new layer and will return an error message.
# This is the correct syntax for adding layers
coverage_plot +
geom_point()
# This will not add the new layer and will return an error message
coverage_plot
+ geom_point()
Building your plots iteratively
Building plots with ggplot2 is typically an iterative process. We start by defining the dataset we’ll use, lay out the axes, and choose a geom:
ggplot(data = variants, aes(x = POS, y = DP)) +
geom_point()
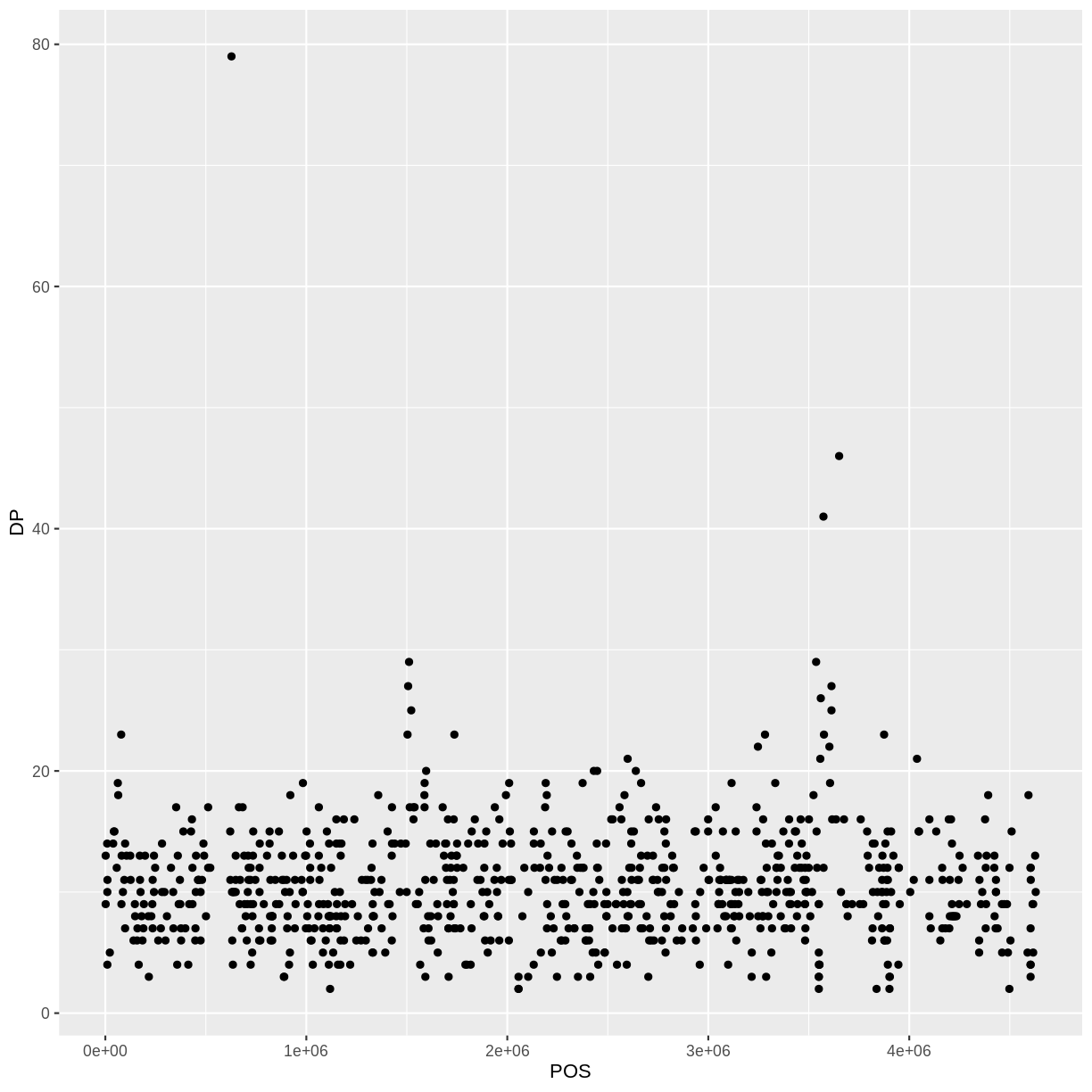
Then, we start modifying this plot to extract more information from it. For instance, we can add transparency (alpha) to avoid over-plotting:
ggplot(data = variants, aes(x = POS, y = DP)) +
geom_point(alpha = 0.5)
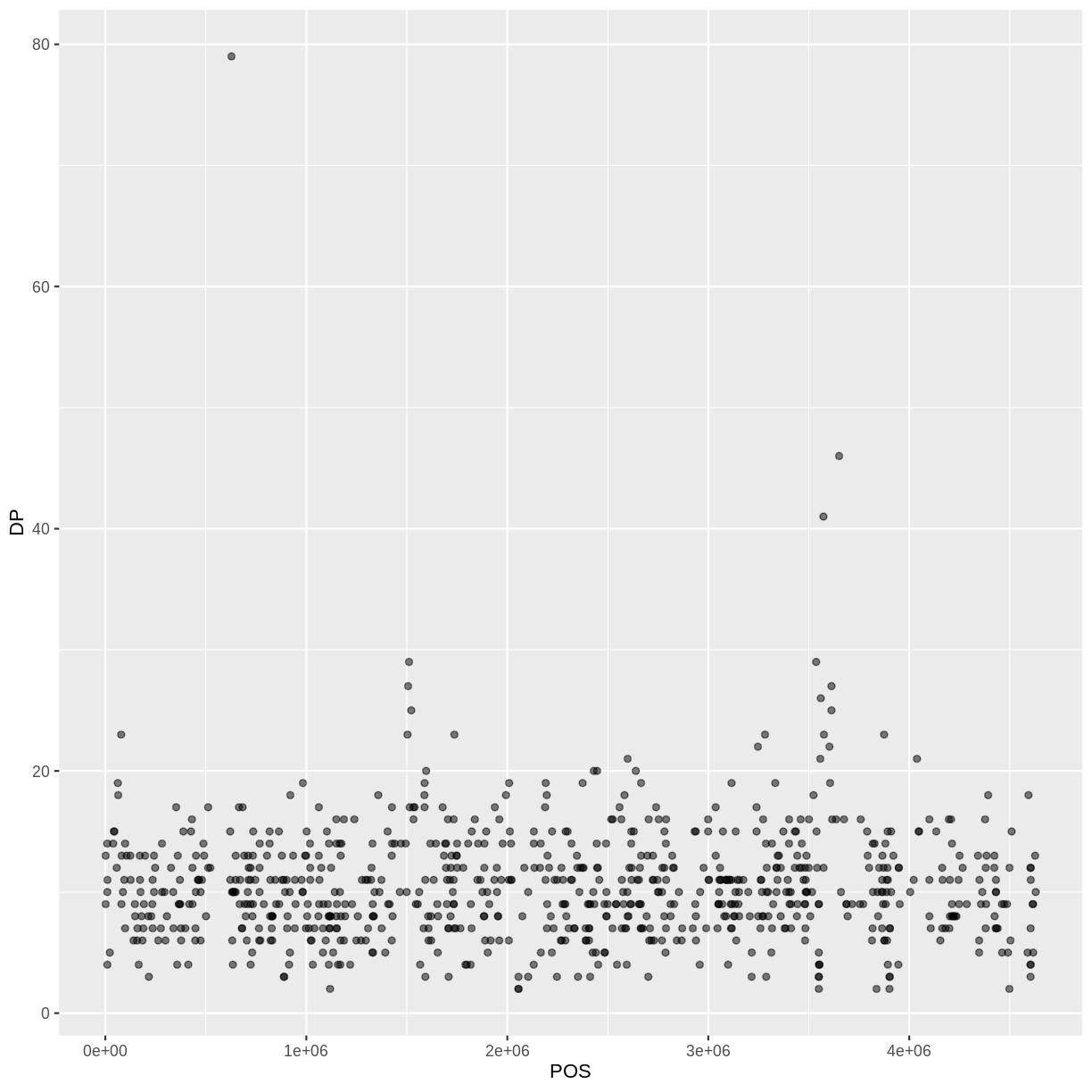
We can also add colors for all the points:
ggplot(data = variants, aes(x = POS, y = DP)) +
geom_point(alpha = 0.5, color = "blue")
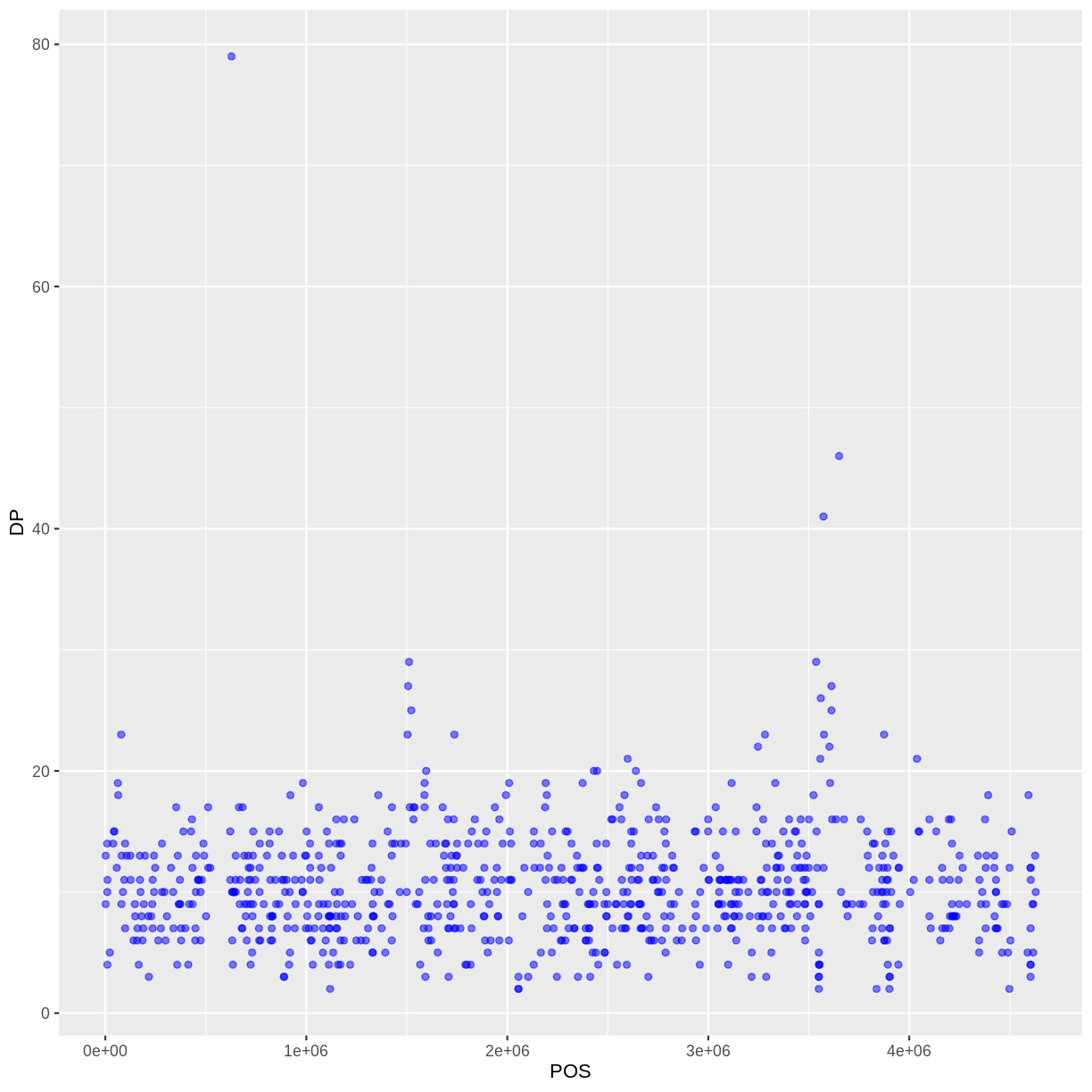
Or to color each species in the plot differently, you could use a vector as an input to the argument color. ggplot2 will provide a different color corresponding to different values in the vector. Here is an example where we color with sample_id:
ggplot(data = variants, aes(x = POS, y = DP, color = sample_id)) +
geom_point(alpha = 0.5)
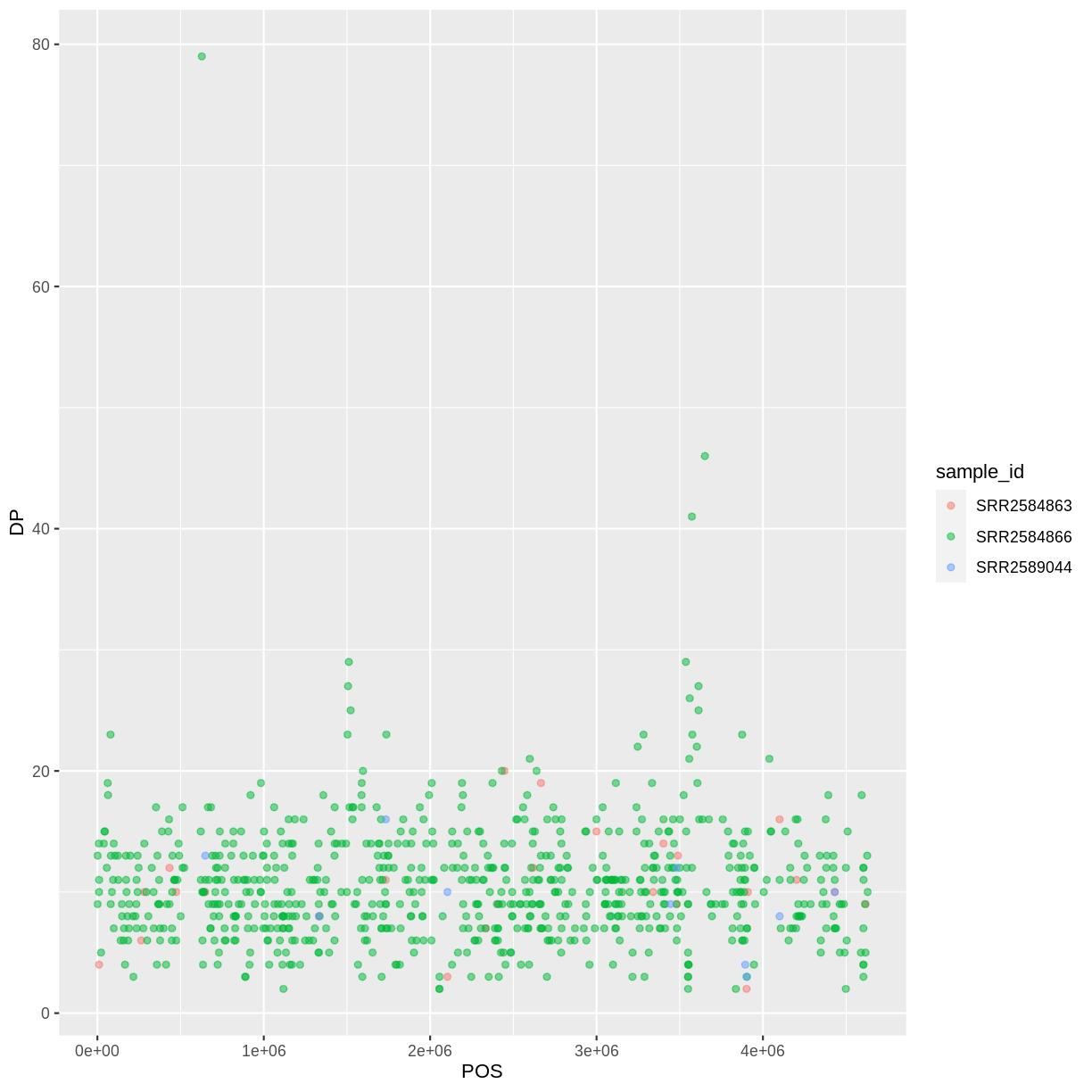
Notice that we can change the geom layer and colors will be still determined by sample_id
ggplot(data = variants, aes(x = POS, y = DP, color = sample_id)) +
geom_point(alpha = 0.5)
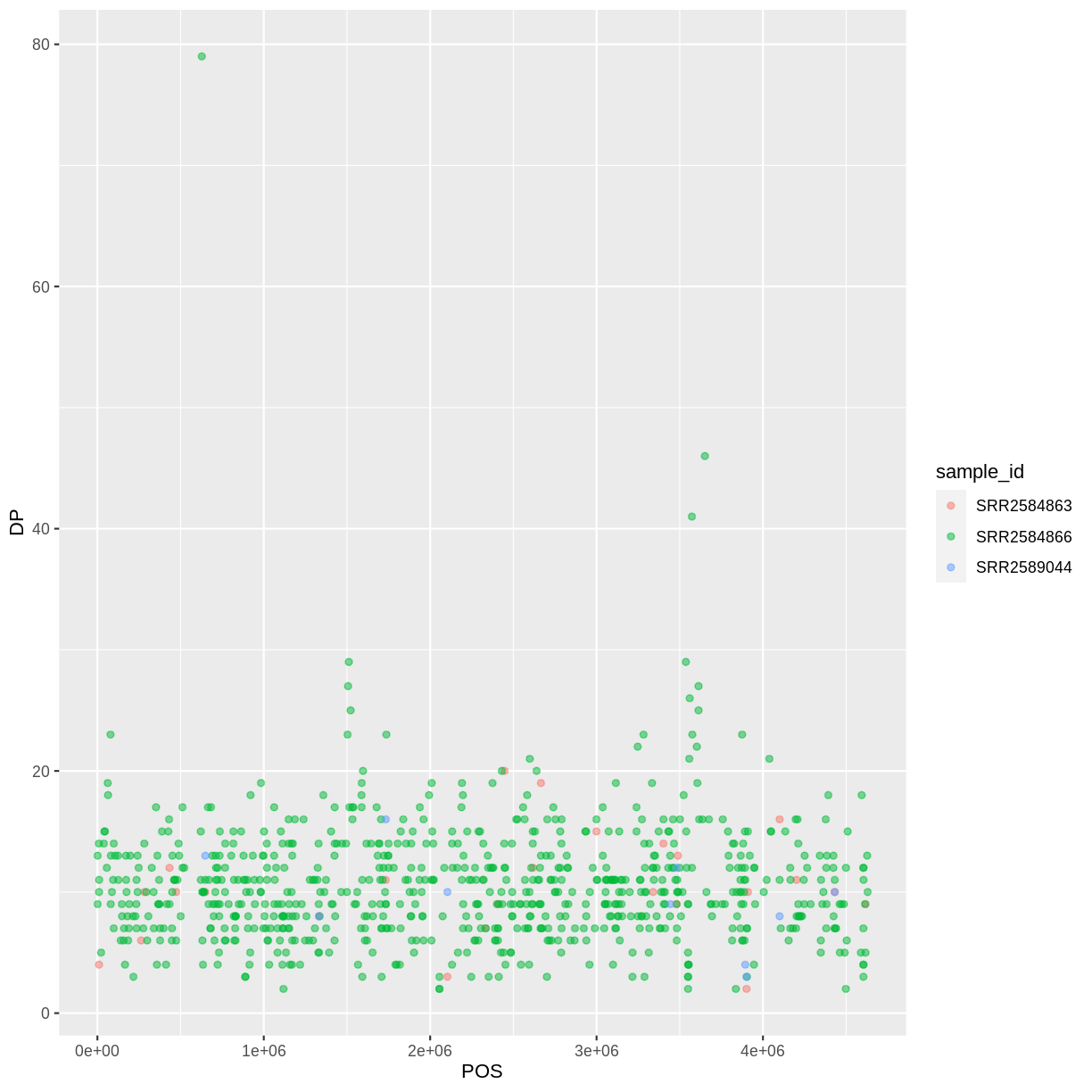
To make our plot more readable, we can add axis labels:
ggplot(data = variants, aes(x = POS, y = DP, color = sample_id)) +
geom_point(alpha = 0.5) +
labs(x = "Base Pair Position",
y = "Read Depth (DP)")
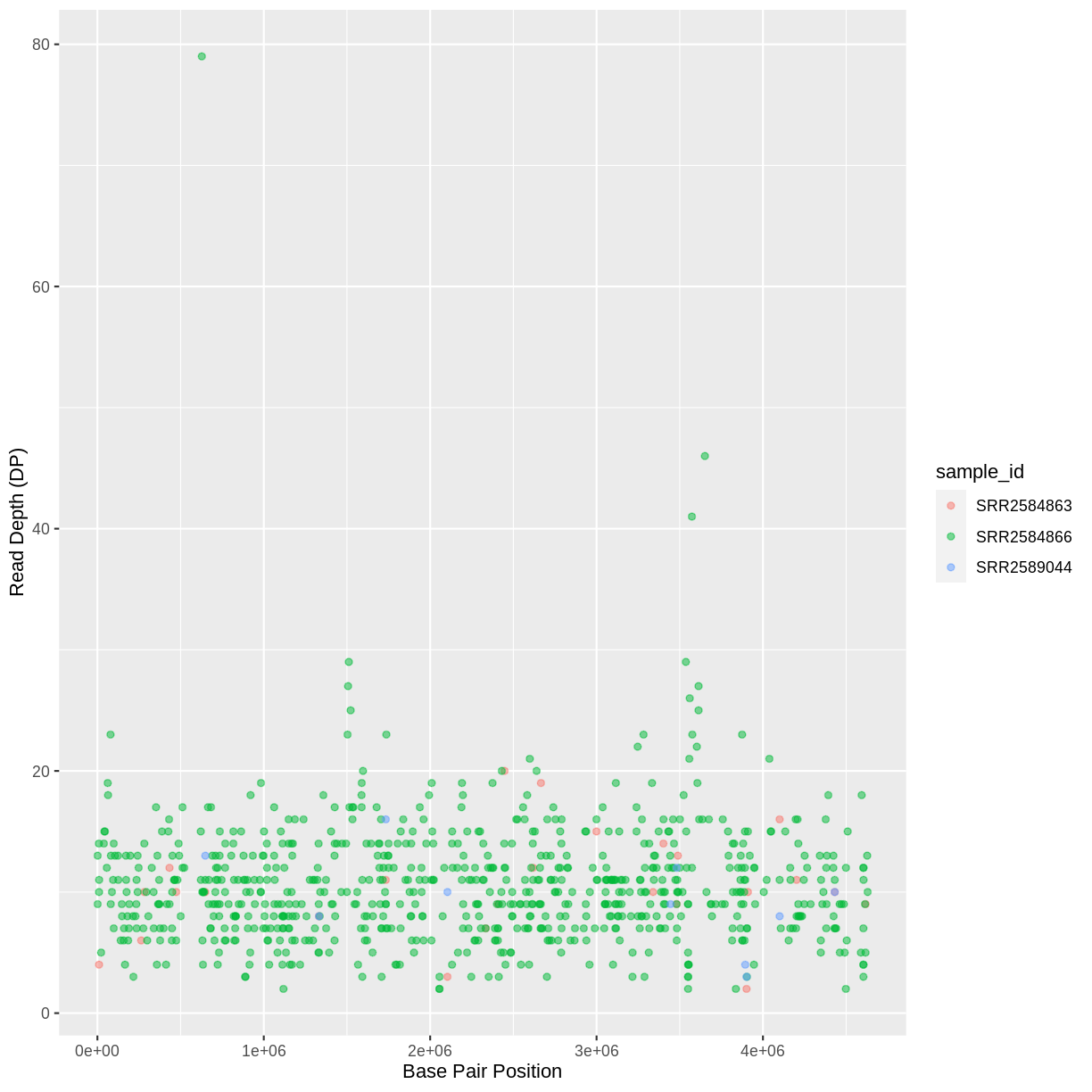
To add a main title to the plot, we use ggtitle():
ggplot(data = variants, aes(x = POS, y = DP, color = sample_id)) +
geom_point(alpha = 0.5) +
labs(x = "Base Pair Position",
y = "Read Depth (DP)") +
ggtitle("Read Depth vs. Position")
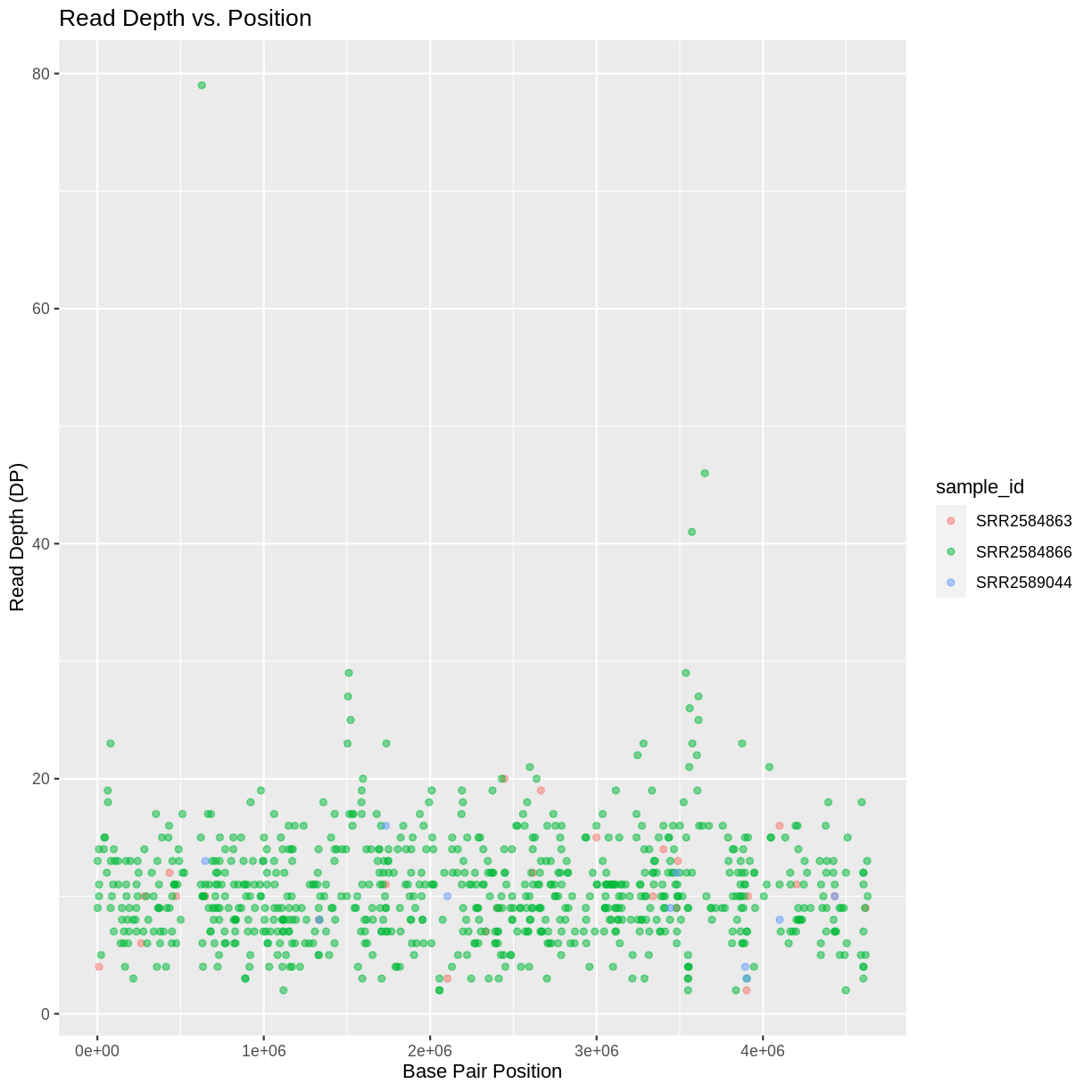
Now the figure is complete and ready to be exported and saved to a file. This can be achieved easily using ggsave(), which can write, by default, the most recent generated figure into different formats (e.g., jpeg, png, pdf) according to the file extension. So, for example, to create a pdf version of the above figure with a dimension of $6\times4$ inches:
ggsave ("depth.pdf", width = 6, height = 4)
If we check the current working directory, there should be a newly created file called depth.pdf with the above plot.
Challenge
Use what you just learned to create a scatter plot of mapping quality (
MQ) over position (POS) with the samples showing in different colors. Make sure to give your plot relevant axis labels.Solution
ggplot(data = variants, aes(x = POS, y = MQ, color = sample_id)) + geom_point() + labs(x = "Base Pair Position", y = "Mapping Quality (MQ)")
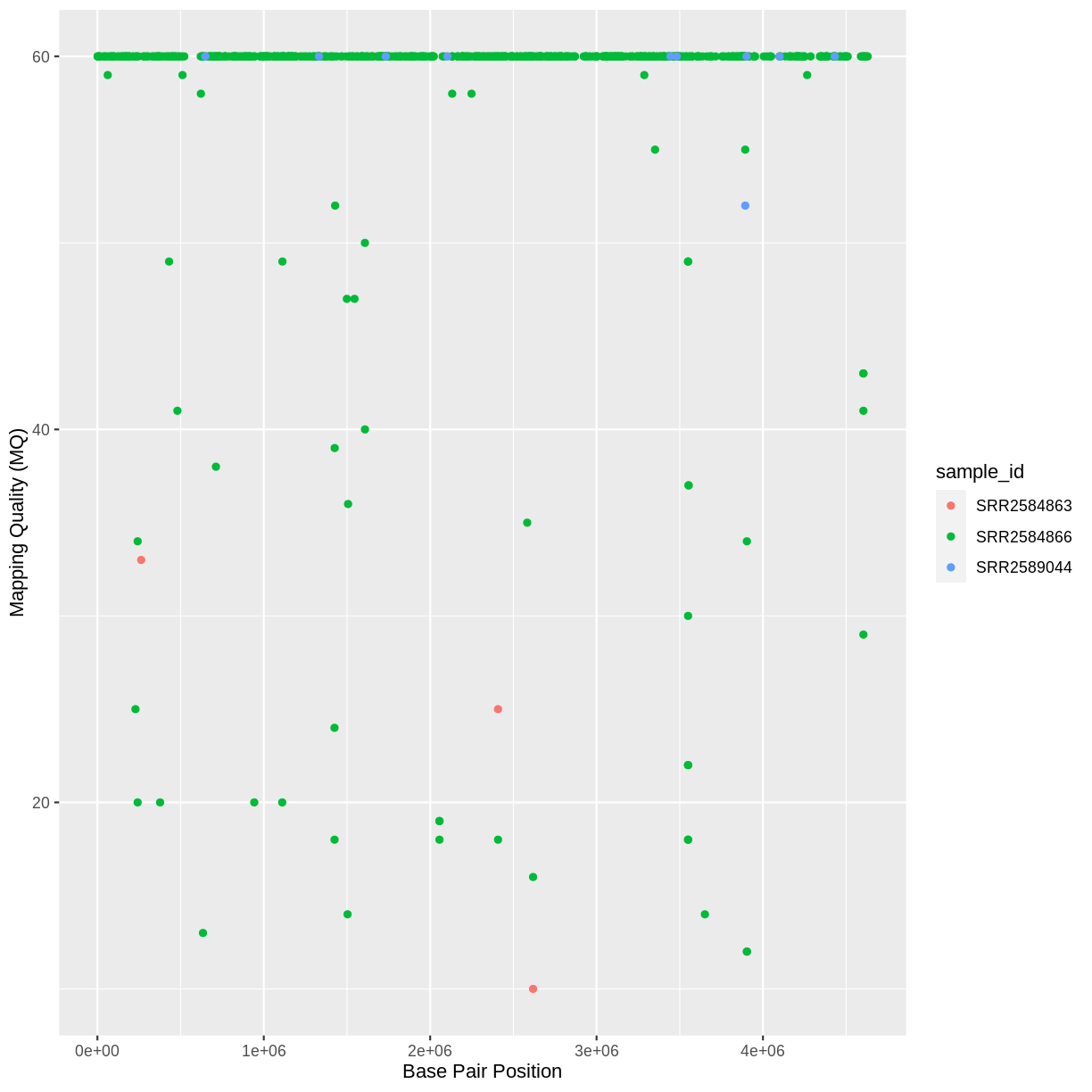
To further customize the plot, we can change the default font format:
ggplot(data = variants, aes(x = POS, y = DP, color = sample_id)) +
geom_point(alpha = 0.5) +
labs(x = "Base Pair Position",
y = "Read Depth (DP)") +
ggtitle("Read Depth vs. Position") +
theme(text = element_text(family = "Bookman"))
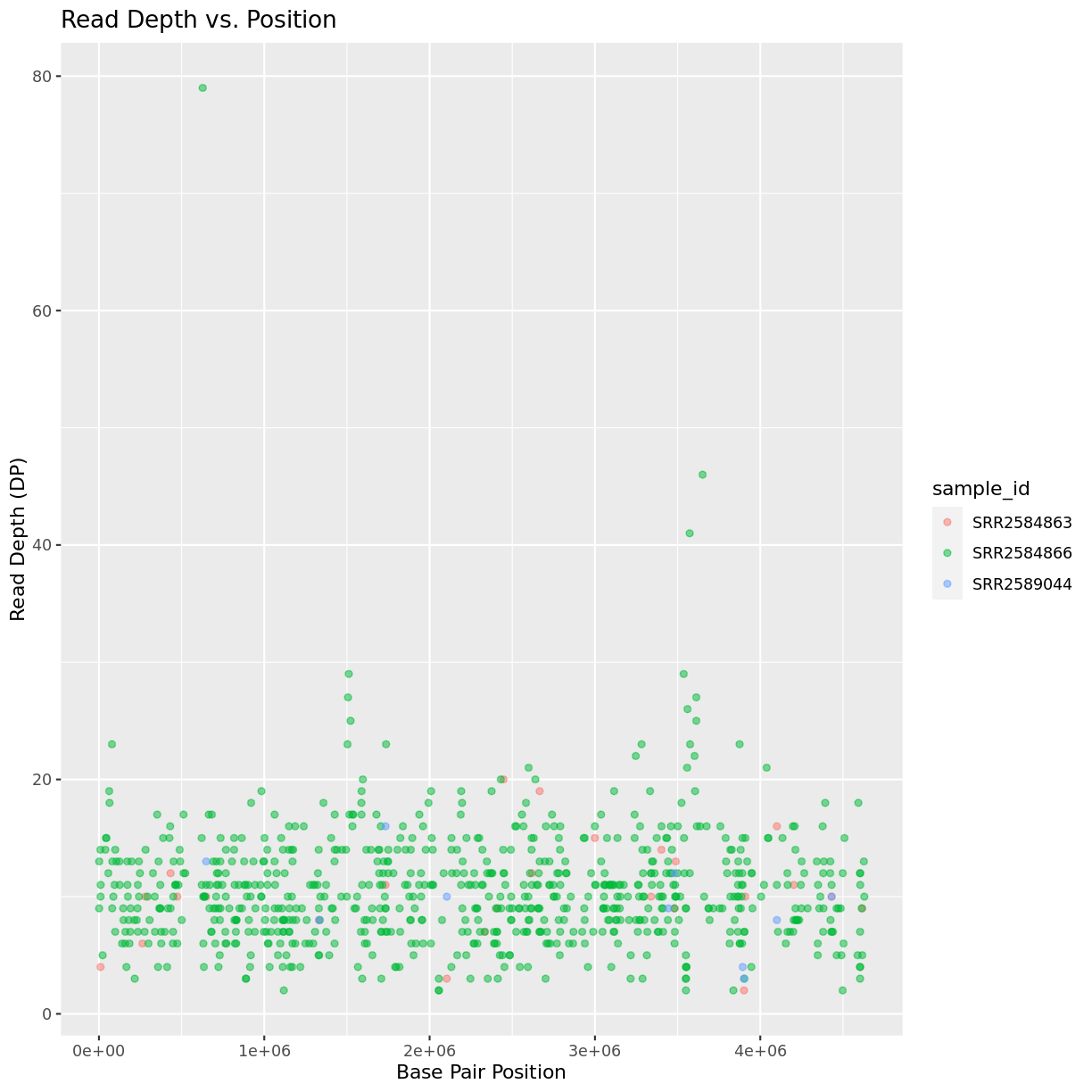
Faceting
ggplot2 has a special technique called faceting that allows the user to split one plot into multiple plots (panels) based on a factor (variable) included in the dataset. We will use it to split our mapping quality plot into three panels, one for each sample.
ggplot(data = variants, aes(x = POS, y = MQ, color = sample_id)) +
geom_point() +
labs(x = "Base Pair Position",
y = "Mapping Quality (MQ)") +
facet_grid(. ~ sample_id)
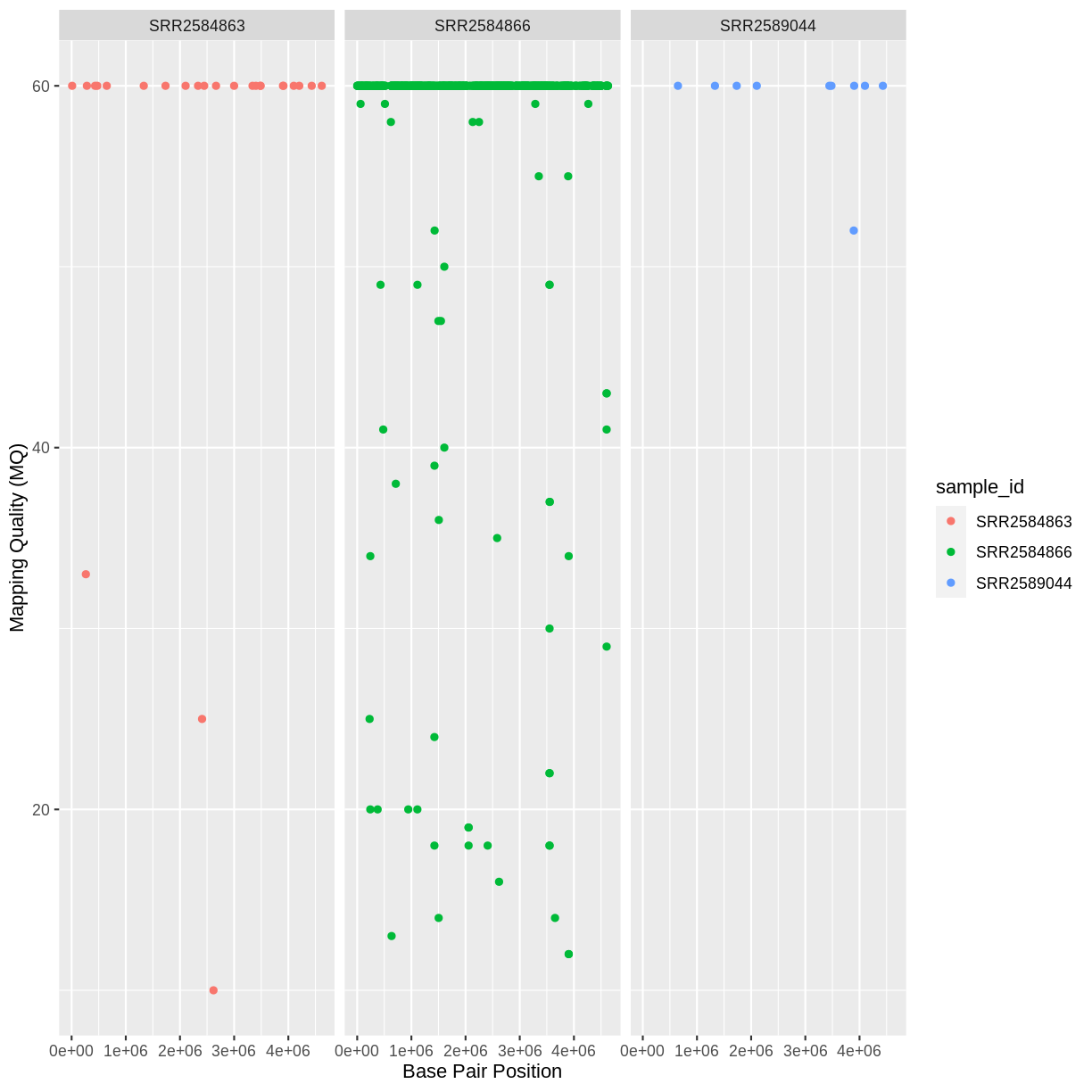
This looks okay, but it would be easier to read if the plot facets were stacked vertically rather than horizontally. The facet_grid geometry allows you to explicitly specify how you want your plots to be arranged via formula notation (rows ~ columns; the dot (.) indicates every other variable in the data i.e., no faceting on that side of the formula).
ggplot(data = variants, aes(x = POS, y = MQ, color = sample_id)) +
geom_point() +
labs(x = "Base Pair Position",
y = "Mapping Quality (MQ)") +
facet_grid(sample_id ~ .)
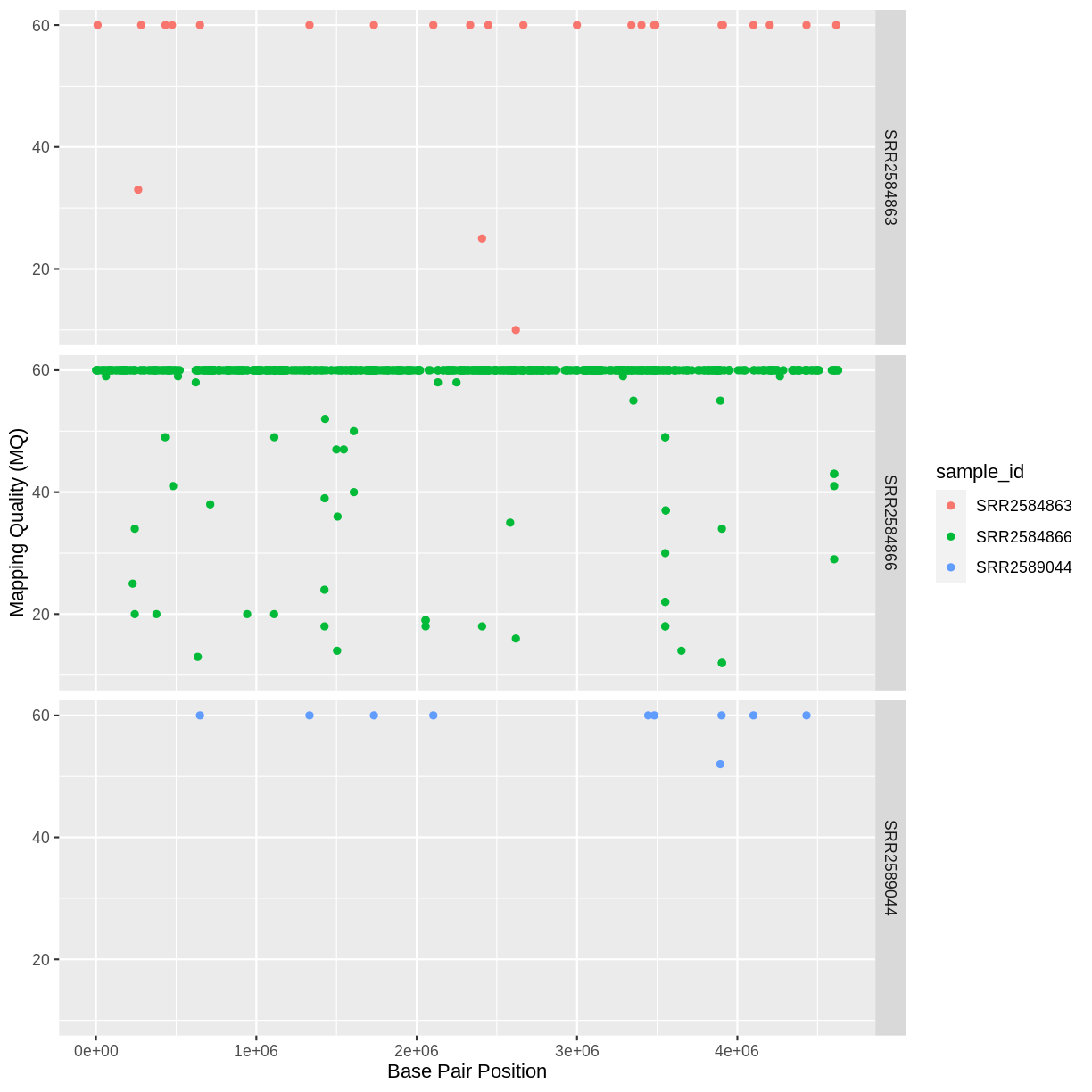
Usually plots with white background look more readable when printed. We can set the background to white using the function theme_bw(). Additionally, you can remove the grid:
ggplot(data = variants, aes(x = POS, y = MQ, color = sample_id)) +
geom_point() +
labs(x = "Base Pair Position",
y = "Mapping Quality (MQ)") +
facet_grid(sample_id ~ .) +
theme_bw() +
theme(panel.grid = element_blank())
Challenge
Use what you just learned to create a scatter plot of PHRED scaled quality (
QUAL) over position (POS) with the samples showing in different colors. Make sure to give your plot relevant axis labels.Solution
ggplot(data = variants, aes(x = POS, y = QUAL, color = sample_id)) + geom_point() + labs(x = "Base Pair Position", y = "PHRED-sacled Quality (QUAL)") + facet_grid(sample_id ~ .)
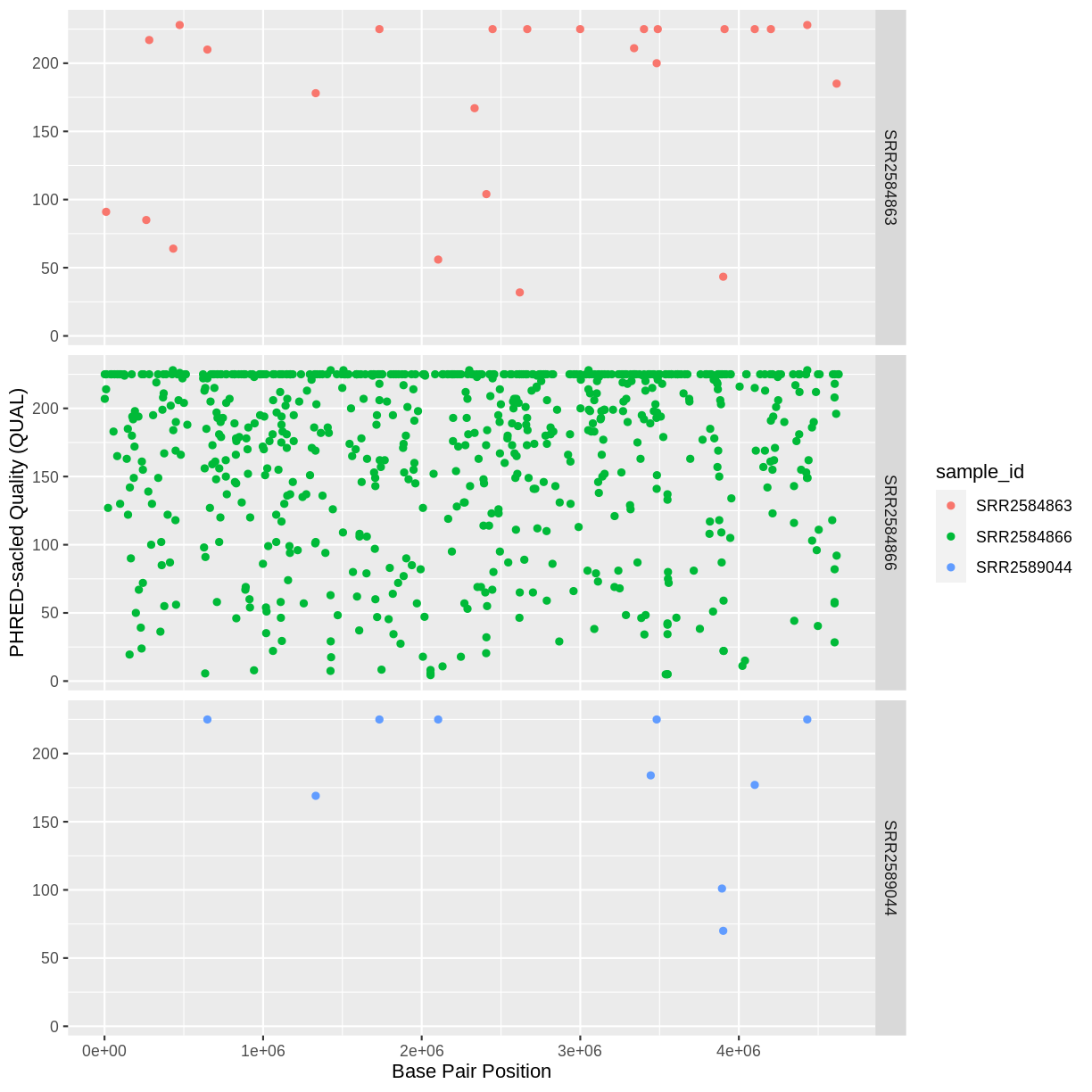
Barplots
We can create barplots using the geom_bar geom. Let’s make a barplot showing the number of variants for each sample that are indels.
ggplot(data = variants, aes(x = INDEL, fill = sample_id)) +
geom_bar() +
facet_grid(sample_id ~ .)
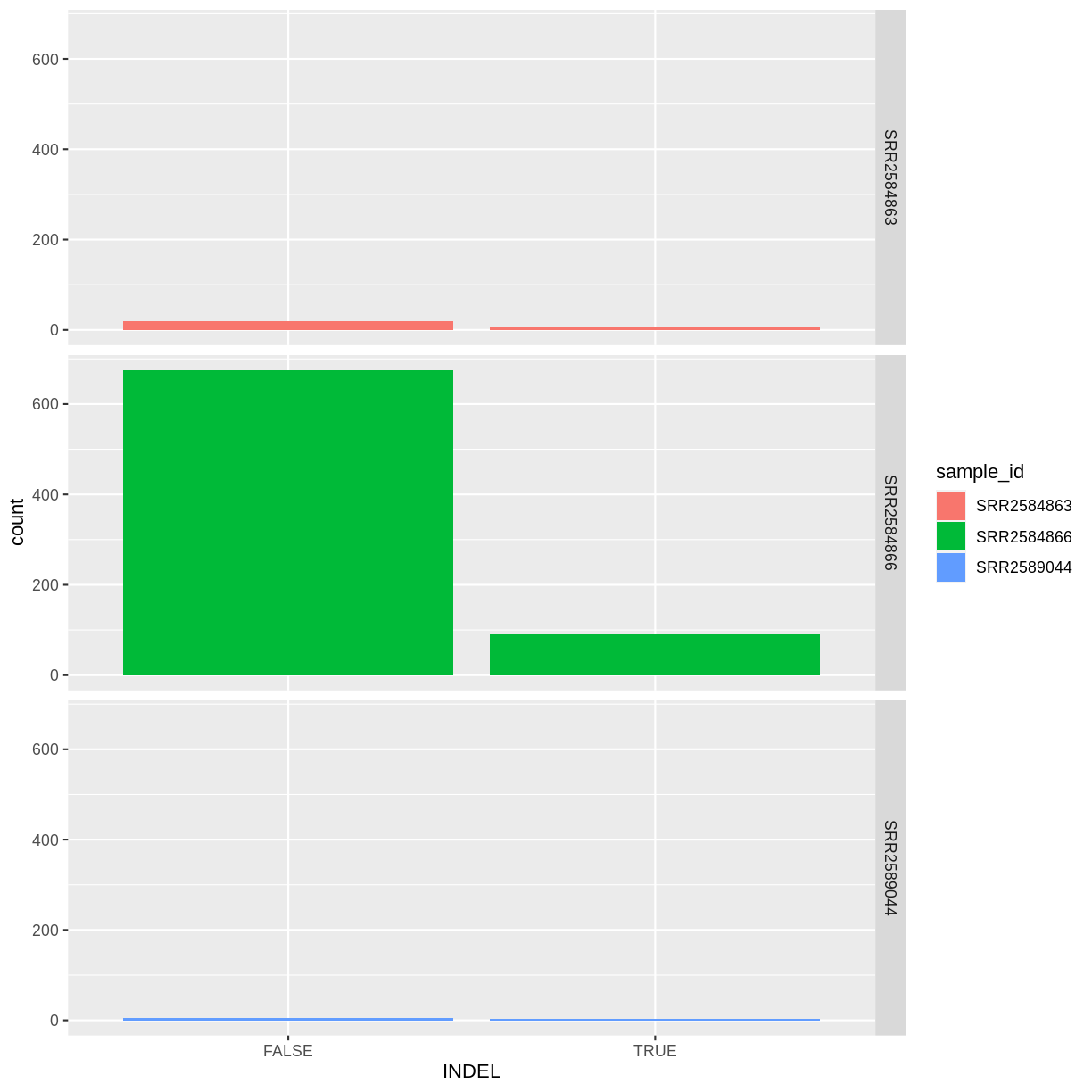
Challenge
Since we already have the sample_id labels on the individual plot facets, we don’t need the legend. Use the help file for
geom_barand any other online resources you want to use to remove the legend from the plot.Solution
ggplot(data = variants, aes(x = INDEL, color = sample_id)) + geom_bar(show.legend = F) + facet_grid(sample_id ~ .)
Density
We can create density plots using the geom_density geom that shows the distribution of of a variable in the dataset. Let’s plot the distribution of DP
ggplot(data = variants, aes(x = DP)) +
geom_density()
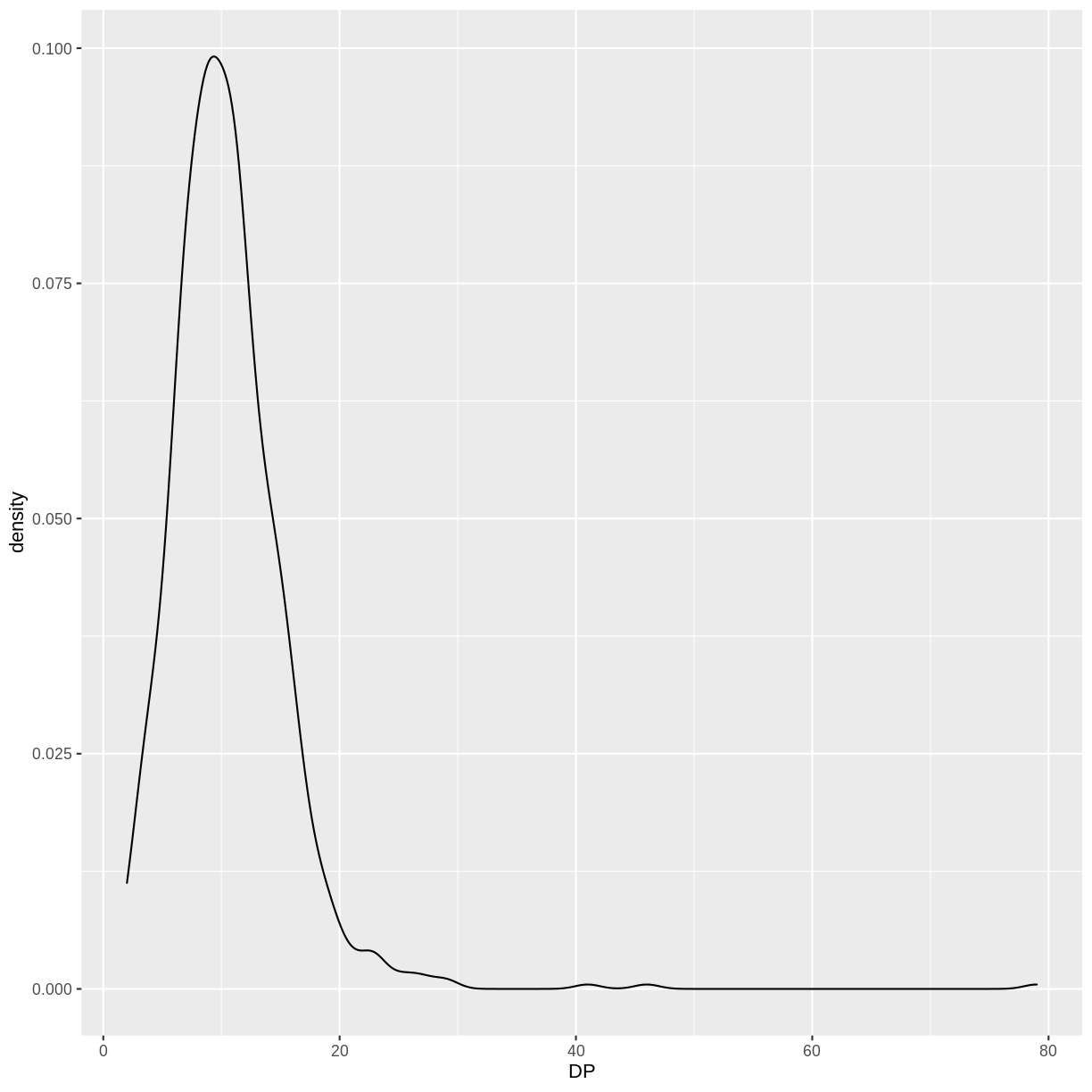
This plot tells us that the most of frequent DP (read depth) for the variants is about 10 reads.
Challenge
Use
geom_densityto plot the distribution ofDPwith a different fill for each sample. Use a white background for the plot.Solution
ggplot(data = variants, aes(x = DP, fill = sample_id)) + geom_density(alpha = 0.5) + theme_bw()
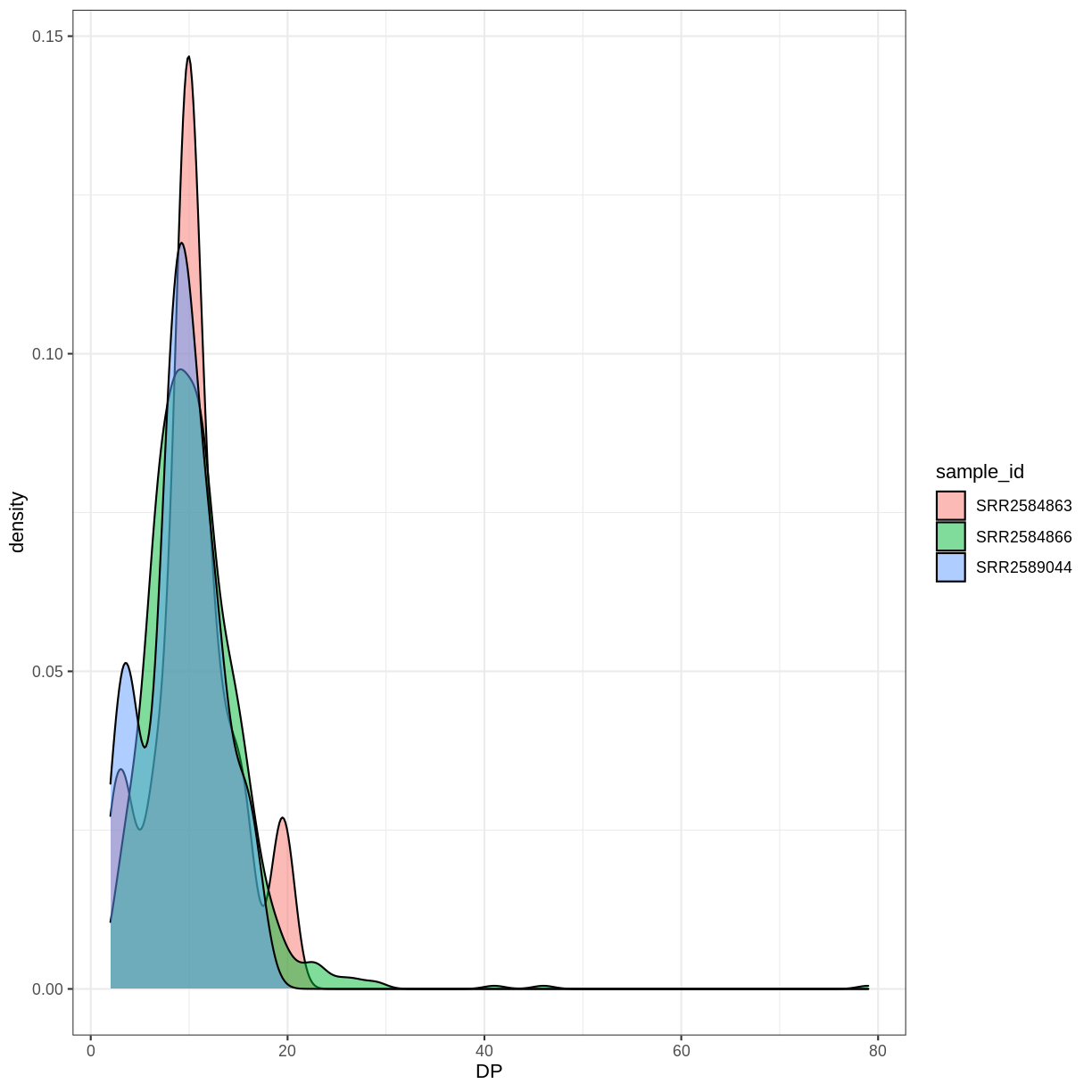
ggplot2 themes
In addition to theme_bw(), which changes the plot background to white, ggplot2 comes with several other themes which can be useful to quickly change the look of your visualization. The complete list of themes is available at https://ggplot2.tidyverse.org/reference/ggtheme.html. theme_minimal() and theme_light() are popular, and theme_void() can be useful as a starting point to create a new hand-crafted theme.
The ggthemes package provides a wide variety of options (including Microsoft Excel, old and new). The ggplot2 extensions website provides a list of packages that extend the capabilities of ggplot2, including additional themes.
Challenge
With all of this information in hand, please take another five minutes to either improve one of the plots generated in this exercise or create a beautiful graph of your own. Use the RStudio
ggplot2cheat sheet for inspiration. Here are some ideas:
- See if you can change the size or shape of the plotting symbol.
- Can you find a way to change the name of the legend? What about its labels?
- Try using a different color palette (see the Cookbook for R.
More ggplot2 Plots
ggplot2 offers many more informative and beautiful plots (geoms) of interest for biologists (although not covered in this lesson) that are worth exploring, such as
geom_tile(), for heatmaps,geom_jitter(), for strip charts, andgeom_violin(), for violin plots
Resources
- ggplot2: Elegant Graphics for Data Analysis (online version)
- The Grammar of Graphics (Statistics and Computing)
- Data Visualization: A Practical Introduction (online version)
- The R Graph Gallery (the book)
Key Points
ggplot2 is a powerful tool for high-quality plots
ggplot2 provides a flexiable and readable grammar to build plots
Producing Reports With knitr
Overview
Teaching: 60 min
Exercises: 15 minQuestions
How can I integrate analyses and reports?
Objectives
Value of reproducible reports
Basics of Markdown
R code chunks
Chunk options
Inline R code
Other output formats
Data analysis reports
Data analysts tend to write a lot of reports, describing their analyses and results, for their collaborators or to document their work for future reference.
When I was first starting out, I’d write an R script with all of my work, and would just send an email to my collaborator, describing the results and attaching various graphs. In discussing the results, there would often be confusion about which graph was which.
I moved to writing formal reports, with Word or LaTeX, but I’d have to spend a lot of time getting the figures to look right. Mostly, the concern is about page breaks.
Everything is easier now that I create a web page (as an html file). It can be one long stream, so I can use tall figures that wouldn’t ordinary fit on one page. Scrolling is your friend.
Literate programming
Ideally, such analysis reports are reproducible documents: If an error is discovered, or if some additional subjects are added to the data, you can just re-compile the report and get the new or corrected results (versus having to reconstruct figures, paste them into a Word document, and further hand-edit various detailed results).
The key tool for R is knitr, which allows you to create a document that is a mixture of text and some chunks of code. When the document is processed by knitr, chunks of R code will be executed, and graphs or other results inserted.
This sort of idea has been called “literate programming”.
knitr allows you to mix basically any sort of text with any sort of code, but we recommend that you use R Markdown, which mixes Markdown with R. Markdown is a light-weight mark-up language for creating web pages.
Creating an R Markdown file
Within R Studio, click File → New File → R Markdown and you’ll get a dialog box like this:

You can stick with the default (HTML output), but give it a title.
Basic components of R Markdown
The initial chunk of text contains instructions for R: you give the thing a title, author, and date, and tell it that you’re going to want to produce html output (in other words, a web page).
---
title: "Initial R Markdown document"
author: "Karl Broman"
date: "April 23, 2015"
output: html_document
---
You can delete any of those fields if you don’t want them included. The double-quotes aren’t strictly necessary in this case. They’re mostly needed if you want to include a colon in the title.
RStudio creates the document with some example text to get you started. Note below that there are chunks like
```{r}
summary(cars)
```
These are chunks of R code that will be executed by knitr and replaced by their results. More on this later.
Also note the web address that’s put between angle brackets (< >) as
well as the double-asterisks in **Knit**. This is
Markdown.
Markdown
Markdown is a system for writing web pages by marking up the text much as you would in an email rather than writing html code. The marked-up text gets converted to html, replacing the marks with the proper html code.
For now, let’s delete all of the stuff that’s there and write a bit of markdown.
You make things bold using two asterisks, like this: **bold**,
and you make things italics by using underscores, like this:
_italics_.
You can make a bulleted list by writing a list with hyphens or asterisks, like this:
* bold with double-asterisks
* italics with underscores
* code-type font with backticks
or like this:
- bold with double-asterisks
- italics with underscores
- code-type font with backticks
Each will appear as:
- bold with double-asterisks
- italics with underscores
- code-type font with backticks
(I prefer hyphens over asterisks, myself.)
You can make a numbered list by just using numbers. You can use the same number over and over if you want:
1. bold with double-asterisks
1. italics with underscores
1. code-type font with backticks
This will appear as:
- bold with double-asterisks
- italics with underscores
- code-type font with backticks
You can make section headers of different sizes by initiating a line
with some number of # symbols:
# Title
## Main section
### Sub-section
#### Sub-sub section
You compile the R Markdown document to an html webpage by clicking the “Knit HTML” in the upper-left. And note the little question mark next to it; click the question mark and you’ll get a “Markdown Quick Reference” (with the Markdown syntax) as well to the RStudio documentation on R Markdown.
Challenge
Create a new R Markdown document. Delete all of the R code chunks and write a bit of Markdown (some sections, some italicized text, and an itemized list).
Convert the document to a webpage.
A bit more Markdown
You can make a hyperlink like this:
[text to show](http://the-web-page.com).
You can include an image file like this: 
You can do subscripts (e.g., F~2~) with F~2 and superscripts (e.g.,
F^2^) with F^2^.
If you know how to write equations in
LaTeX, you’ll be glad to know that
you can use $ $ and $$ $$ to insert math equations, like
$E = mc^2$ and
$$y = \mu + \sum_{i=1}^p \beta_i x_i + \epsilon$$
R code chunks
Markdown is interesting and useful, but the real power comes from mixing markdown with chunks of R code. This is R Markdown. When processed, the R code will be executed; if they produce figures, the figures will be inserted in the final document.
The main code chunks look like this:
```{r load_data}
gapminder <- read.csv("~/Desktop/gapminder.csv")
```
That is, you place a chunk of R code between ```{r chunk_name}
and ```. It’s a good idea to give each chunk
a name, as they will help you to fix errors and, if any graphs are
produced, the file names are based on the name of the code chunk that
produced them.
Challenge
Add code chunks to
- Load the ggplot2 package
- Read the gapminder data
- Create a plot
How things get compiled
When you press the “Knit HTML” button, the R Markdown document is processed by knitr and a plain Markdown document is produced (as well as, potentially, a set of figure files): the R code is executed and replaced by both the input and the output; if figures are produced, links to those figures are included.
The Markdown and figure documents are then processed by the tool pandoc, which converts the Markdown file into an html file, with the figures embedded.
Chunk options
There are a variety of options to affect how the code chunks are treated.
- Use
echo = FALSEto avoid having the code itself shown. - Use
results = "hide"to avoid having any results printed. - Use
eval = FALSEto have the code shown but not evaluated. - Use
warning = FALSEandmessage = FALSEto hide any warnings or messages produced. - Use
fig.heightandfig.widthto control the size of the figures produced (in inches).
So you might write:
```{r load_libraries, echo=FALSE, message=FALSE}
library("dplyr")
library("ggplot2")
```
Often there will be particular options that you’ll want to use repeatedly; for this, you can set global chunk options, like so:
```{r global_options, echo=FALSE}
knitr::opts_chunk$set(fig.path="Figs/", message=FALSE, warning=FALSE,
echo=FALSE, results="hide", fig.width=11)
```
The fig.path option defines where the figures will be saved. The /
here is really important; without it, the figures would be saved in
the standard place but just with names that being with Figs.
If you have multiple R Markdown files in a common directory, you might
want to use fig.path to define separate prefixes for the figure file
names, like fig.path="Figs/cleaning-" and fig.path="Figs/analysis-".
Challenge
Use chunk options to control the size of a figure and to hide the code.
Inline R code
You can make every number in your report reproducible. Use
`r and ` for an in-line code chunk,
like so: `r round(some_value, 2)`. The code will be
executed and replaced with the value of the result.
Don’t let these in-line chunks get split across lines.
Perhaps precede the paragraph with a larger code chunk that does
calculations and defines things, with include=FALSE for that larger
chunk (which is the same as echo=FALSE and results="hide").
I’m very particular about rounding in such situations. I may want
2.0, but round(2.03, 1) will give just 2.
The
myround
function in the broman package handles
this.
Challenge
Try out a bit of in-line R code.
Other output options
You can also convert R Markdown to a PDF or a Word document. Click the
little triangle next to the “Knit HTML” button to get a drop-down
menu. Or you could put pdf_document or word_document in the header
of the file.
Tip: Creating PDF documents
Creating .pdf documents may require installation of some extra software. If required this is detailed in an error message.
Tex for windows is available here.
Tex for mac is available here.
Resources
- Knitr in a knutshell tutorial
- Dynamic Documents with R and knitr (book)
- R Markdown documentation
- R Markdown cheat sheet (PDF)
- Getting started with R Markdown
- Reproducible Reporting
- Introducing Bookdown
Key Points
Keep reporting and R software together in one document using R Markdown.
Control formatting using chunk options.
knitrcan convert R Markdown documents to PDF and other formats.
Getting help with R
Overview
Teaching: 10 min
Exercises: 5 minQuestions
How do I get help using R and RStudio?
Objectives
Locate help for an R function using
?,??, andargs()Check the version of R
Be able to ask effective questions when searching for help on forums or using web searches
Getting help with R

No matter how much experience you have with R, you will find yourself needing help. There is no shame in researching how to do something in R, and most people will find themselves looking up how to do the same things that they “should know how to do” over and over again. Here are some tips to make this process as helpful and efficient as possible.
“Never memorize something that you can look up” – A. Einstein
Finding help on Stackoverflow and Biostars
Two popular websites will be of great help with many R problems. For general R questions, Stack Overflow is probably the most popular online community for developers. If you start your question “How to do X in R” results from Stack Overflow are usually near the top of the list. For bioinformatics specific questions, Biostars is a popular online forum.
Tip: Asking for help using online forums:
- When searching for R help, look for answers with the r tag.
- Get an account; not required to view answers but to required to post
- Put in effort to check thoroughly before you post a question; folks get annoyed if you ask a very common question that has been answered multiple times
- Be careful. While forums are very helpful, you can’t know for sure if the advice you are getting is correct
- See the How to ask for R help blog post for more useful tips
Help people help you
Often, in order to duplicate the issue you are having, someone may need to see the data you are working with or verify the versions of R or R packages you are using. The following R functions will help with this:
You can check the version of R you are working with using the sessionInfo()
function. Actually, it is good to save this information as part of your notes
on any analysis you are doing. When you run the same script that has worked fine
a dozen times before, looking back at these notes will remind you that you
upgraded R and forget to check your script.
sessionInfo()
R version 3.2.3 (2015-12-10) Platform: x86_64-pc-linux-gnu (64-bit) Running under: Ubuntu 14.04.3 LTS locale: [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C LC_TIME=en_US.UTF-8 [4] LC_COLLATE=en_US.UTF-8 LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8 [7] LC_PAPER=en_US.UTF-8 LC_NAME=C LC_ADDRESS=C [10] LC_TELEPHONE=C LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C attached base packages: [1] stats graphics grDevices utils datasets methods base loaded via a namespace (and not attached): [1] tools_3.2.3 packrat_0.4.9-1
Many times, there may be some issues with your data and the way it is formatted.
In that case, you may want to share that data with someone else. However, you
may not need to share the whole dataset; looking at a subset of your 50,000 row,
10,000 column dataframe may be TMI (too much information)! You can take an
object you have in memory such as dataframe (if you don’t know what this means
yet, we will get to it!) and save it to a file. In our example we will use the
dput() function on the iris dataframe which is an example dataset that is
installed in R:
dput(head(iris)) # iris is an example data.frame that comes with R # the `head()` function just takes the first 6 lines of the iris dataset
This generates some output (below) which you will be better able to interpret after covering the other R lessons. This info would be helpful in understanding how the data is formatted and possibly revealing problematic issues.
structure(list(Sepal.Length = c(5.1, 4.9, 4.7, 4.6, 5, 5.4), Sepal.Width = c(3.5, 3, 3.2, 3.1, 3.6, 3.9), Petal.Length = c(1.4, 1.4, 1.3, 1.5, 1.4, 1.7), Petal.Width = c(0.2, 0.2, 0.2, 0.2, 0.2, 0.4), Species = structure(c(1L, 1L, 1L, 1L, 1L, 1L), .Label = c("setosa", "versicolor", "virginica"), class = "factor")), .Names = c("Sepal.Length", "Sepal.Width", "Petal.Length", "Petal.Width", "Species"), row.names = c(NA, 6L), class = "data.frame")
Alternatively, you can also save objects in R memory to a file by specifying
the name of the object, in this case the iris data frame, and passing a
filename to the file= argument.
saveRDS(iris, file="iris.rds") # By convention, we use the .rds file extension
Final FAQs on R
Finally, here are a few pieces of introductory R knowledge that are too good to pass up. While we won’t return to them in this course, we put them here because they come up commonly:
Do I need to click Run every time I want to run a script?
- No. In fact, the most common shortcut key allows you to run a command (or
any lines of the script that are highlighted):
- Windows execution shortcut: Ctrl+Enter
- Mac execution shortcut: Cmd(⌘)+Enter
To see a complete list of shortcuts, click on the Tools menu and select Keyboard Shortcuts Help
What’s with the brackets in R console output?
- R returns an index with your result. When your result contains multiple values, the number tells you what ordinal number begins the line, for example:
1:101 # generates the sequence of numbers from 1 to 101
[1] 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
[19] 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36
[37] 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54
[55] 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72
[73] 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90
[91] 91 92 93 94 95 96 97 98 99 100 101
In the output above, [81] indicates that the first value on that line is the
81st item in your result
Can I run my R script without RStudio?
- Yes, remember - RStudio is running R. You get to use lots of the enhancements RStudio provides, but R works independent of RStudio. See these tips for running your commands at the command line
Where else can I learn about RStudio?
- Check out the Help menu, especially “Cheatsheets” section
Key Points
R provides thousands of functions for analyzing data, and provides several way to get help
Using R will mean searching for online help, and there are tips and resources on how to search effectively